
B Cells Anonymous
B Cells do not stem from a singular, centralized "immune response." For this reason, Francis's obsession never made sense to begin with. (OAS lit review supplement.)

Reader beware: The following is both a nitty-gritty science post, and is being published in something of a draft / Beta version.

The previous post discussed what was known about flu and “antibodies” up to 1953. What about our current understanding of how the immune system forms antibodies?
As Part 2 of the Official Unglossed “OAS Lit Review” will be focused on the modern “evidence” for OAS, a purely mechanistic argument against the myth doesn’t have any neat fit within the series proper. So, this entry may be thought of either as a supplement to the review series or as a stand-alone post.
As with Part 1, the reader is encouraged to sit this one out if the docket is not appealing. (Strikethrough portions will be added in a later update):
How B Cell “memory” is not static, but distributed in space and time
What Thomas Francis observed
How should Francis’s observations be interpreted, if considered today?
How is “immune imprinting” any different than the current accepted model of B Cell memory immunity? (It’s not.)
Once again, a changelog for updates will be kept in the footnotes if needed.1
1. B Cell “memory” is not static, but distributed in space and time
Boot Camp
Focusing exclusively on the first-time development of a B Cell memory response still leaves us confronting a bewildering jungle of activity. But beholding the entirety is not necessary to grasp the central point, and so this discussion will be maximally minimalist. And so here is the process when a novel pathogen is encountered somewhere in the body:2

Elements of the “circuitry” not involving B Cells, like Antigen Presentation Cells, are only minimally portrayed. Important points:
In the case of a novel antigen, naive B Cell encounter will be accompanied by IgM and a complement protein. This behaves like a “tag” which tells the B Cell to get to work.
This leads to both short-term responses (IgM antibodies), and a longer-term response which is more strictly regulated by T Cells - this design is both to achieve a higher-affinity B Cell / antibody model for long term protection, and to guard against auto-immunity. Long-lived memory T Cells are formed along the way.
The final lineage of plasma and memory B Cells from a given Germinal Center may last a lifetime, but the Germinal Center does not. Any future long-term B Cell enrichment against this antigen or a related antigen on the same scale3 will require starting the process over again, and creating a new generation of long-lived etc.
Antibodies eventually fade from acute phase levels (not pictured). This is important since even a “good” antibody probably has some self-reactivity, especially in the context of viruses which so closely mimic our own designs.
This also implicitly creates a bigger window for initiation of novel memory immune responses upon re-encounter. More below.
In the case of a symptomatic infection, it can be expected that multiple germinal centers will arise, either in the same lymph organ or in multiple. These can all produce different B and T Cell responses and different antibodies against different antigens. The immune response is always distributed, which is why real viral encounter creates an entire spectrum of memory immune response.
More discussion about the whole Germinal Center “deal” is at “The 60 Day RNA Mystery, Pt 1.”
Software Update
If the same agent - let’s say a virus - reappears, existing IgG antibody will bind to it, but may not be present in high enough amounts to suppress early replication. However, Memory B Cells are ever ready to ramp up production. Between this, innate immunity, and T Cell recognition, the virus’s prospects are grim.
Now let’s say a mutant version of the virus - a new iteration of flu virus, for example - is up to mischief. What will happen then?
This turns out to come down to the lowly IgM + complement protein combination that started things off last time. Naive B Cells that encounter an antigen tightly bound with IgG will not be stimulated - leaving the old memory immune response to dominate - whereas those that encounter antigen bound with IgM + complement protein will kick into gear.4 And so the answer is actually up to the virus / antigen:
If it is well-enough bound by the previous antibodies, there will not be a new long-term immune response.
If it is not, then generic IgM will out-compete it in some cases and will prompt a new long-term immune response which, because it will take place in a newly-formed Germinal Center, must be based on the new version of the antigen.
And so even if, like us, the immune system would prefer not to update every time a new version of a virus comes along, if previous IgG antibody cannot do the job then it will not have a choice. The central B Cell “logic” portion is adapted from the model in Kim, D. et al.:5

And so, if we are asking the question:
What does the present understanding predict will happen when the immune system meets a more-than-minimally different version of a prior antigen?
The answer is:
Previous antibodies will be ramped up, and new antibodies will be formed. The magnitude of the latter will depend on how well the previous antibodies bind.
However, there is absolutely no room in our understanding of the immune system to propose that previous antibodies should not ramp up. The new version of the antigen will still stimulate Memory B Cells whose receptors partially bind it.
In 1953, Thomas Francis didn’t know what a Memory B Cell was, let alone anything else in our circuit. So his opinion on antibodies is fundamentally uninformed.
And yet note the elegant (though hypothetical) feedback mechanism I have included in my illustration. Just as surely as a weakly-binding variant antigen will both stimulate Memory B Cell expansion and a novel Germinal Center response, new short-lived Plasma Cells will emerge with “prototype” versions of IgM antibodies for the new antigen. These (hypothetically) will compete with the old IgG model antibodies, potentially tipping the scale away from old-model ramp-up and toward even more Germinal Center activation. It’s a thought, anyway.
In either case, the worse a previous antibody binds, the more stimulation there will be for a novel response. This implicit balance is elegant enough:
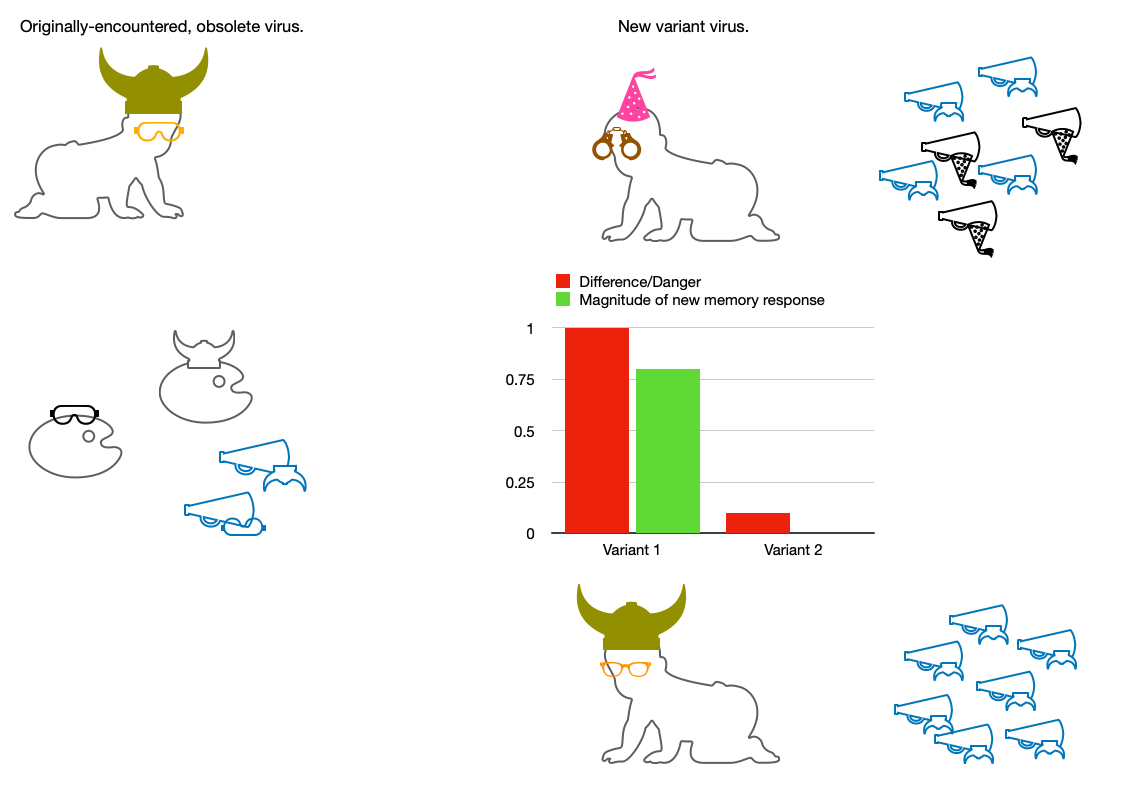
The new Germinal Center response is both made necessary by the novelty of the antigen, and by necessity will be geared to the new version. This is the only way that Germinal Centers can work: The antigen is copied from the virus that is in the body now, not from a previous antibody model.
The suggestion, in the context of SARS-CoV-2, that the Memory B Cells and Plasma Cells generated in reply to the Covid vaccine-induced spike proteins can somehow forbid the body from initiating a new immune memory response in any Germinal Center, anywhere, is without merit. And that new memory response would have to be to whatever the immune system is actually encountering: i.e., the new variant of the virus.
This is true whether for the N Protein of the virus.
Or for the Spike protein.
The Covid vaccinated, whatever else their problems are (like taking an experimental genetic intervention over and over because the news scares them), are able to adapt along with SARS-CoV-2.
Naturally, there are other considerations, the immune system being such a vast and poorly understood apparatus. Among which:
To what extent is Germinal Center affinity maturation more prolonged in initial encounter vs. later encounters, particularly with regard to the flu HA protein, absent significant viral load? (It would seem that this difference explains why animal flu vaccination can produce such stronger results the first time.)
To what extent does this contribute to lifelong cross-strain immunity?
How bad the problem of immunosenescence (novel immune responses will diminish in magnitude regardless of the “balance” in new B Cell stimulation).
But once again, how beneficial the tradeoff in prior immune memory?
etc.
These questions may not be answerable. But the burden is on OAS to somehow do so - to prove that re-encounter with flu at later stages of life on balance goes worse because of cross-protection than it would if we didn’t retain immune memory according to the understood system.
2. What Francis saw
Astoundingly, the paper that Francis and two underlings assembled which first established a foothold for his pet theory was all about pooled blood. (Note that the significance of the depicted strains is better understood after a full visit to the contemporary flu research “scene,” in Part 1.)

The findings:6
Blood for adult donors (processed into gamma globulin) increased in binding vs. PR8 (Francis’ prototypical 1934 flu strain) after 1947, when “A Prime” (the antigenic shift that made PR8 obsolete) appeared on the scene. Exactly how the samples for 1947 were classified (“pre” or “post”?) is unclear. It hardly matters.

The use of pooled blood (and mathematical blending of multiple years!) means that there is no way to distinguish between individually raised levels of immune recognition (some people got more antibodies against PR8) vs. raised levels of individual immune recognition (more people got some antibodies against PR8). It’s the worst way to measure “immunity” possible. The reader may assess for themselves whether Francis and co. seem to even understand this fundamental flaw in their approach; I am far from convinced.
If blood is pooled by age, different age groups demonstrate higher binding against different strains. Again, there is no way to parse whether this is due to donors recognizing the strains more strongly or more donors recognizing the strains.
Shope’s “Swine” (the 1918 flu stand-in) recognition is strong in pooled blood for adults who were alive around that time.
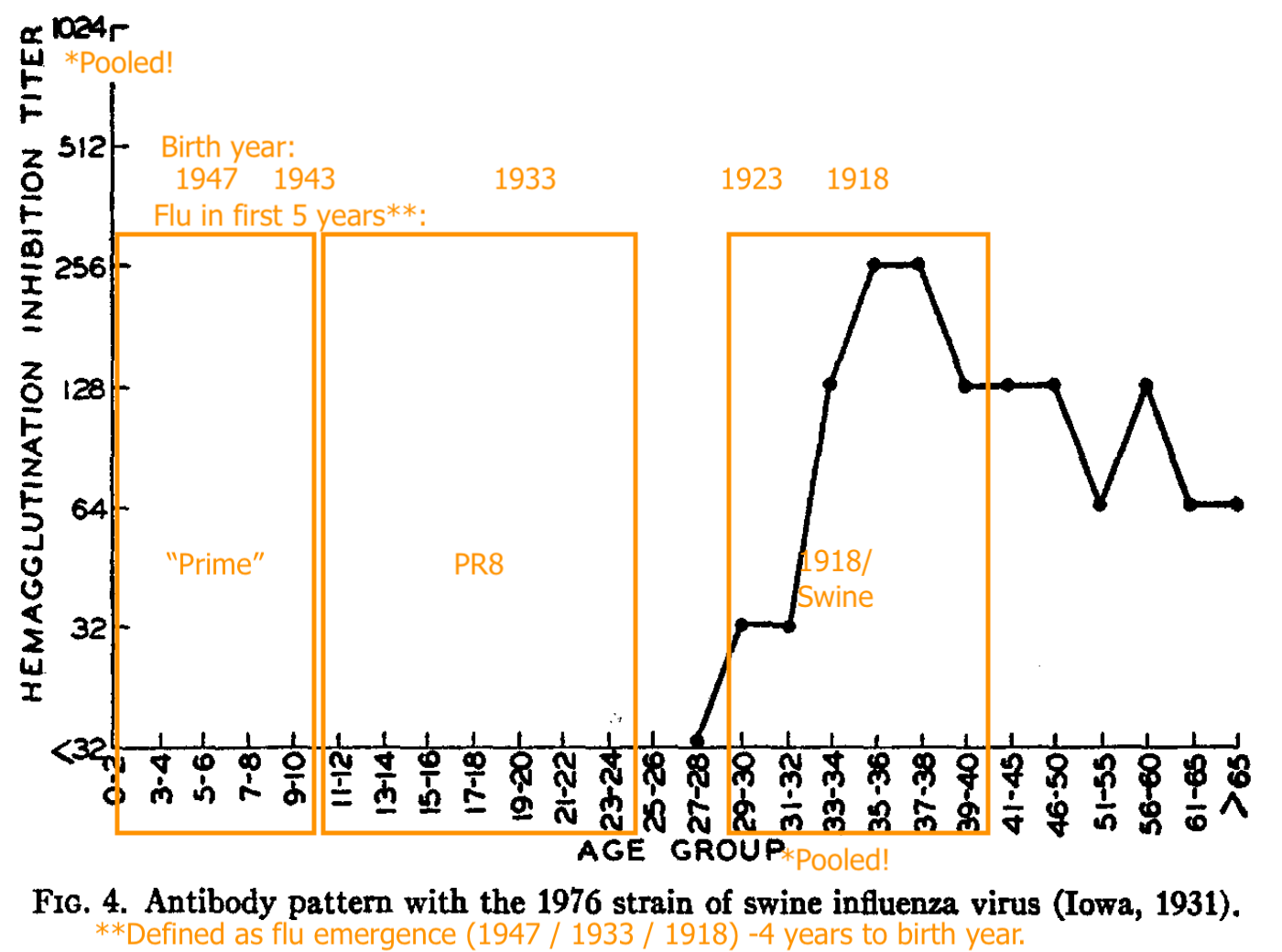
PR8 (1934 - 43) recognition is absent in pooled young children, strong for pooled older teeens and young adults, less strong for pooled adults born in 1923 or before (1918 - 22 being the likely span of the 1918 flu’s dominance).
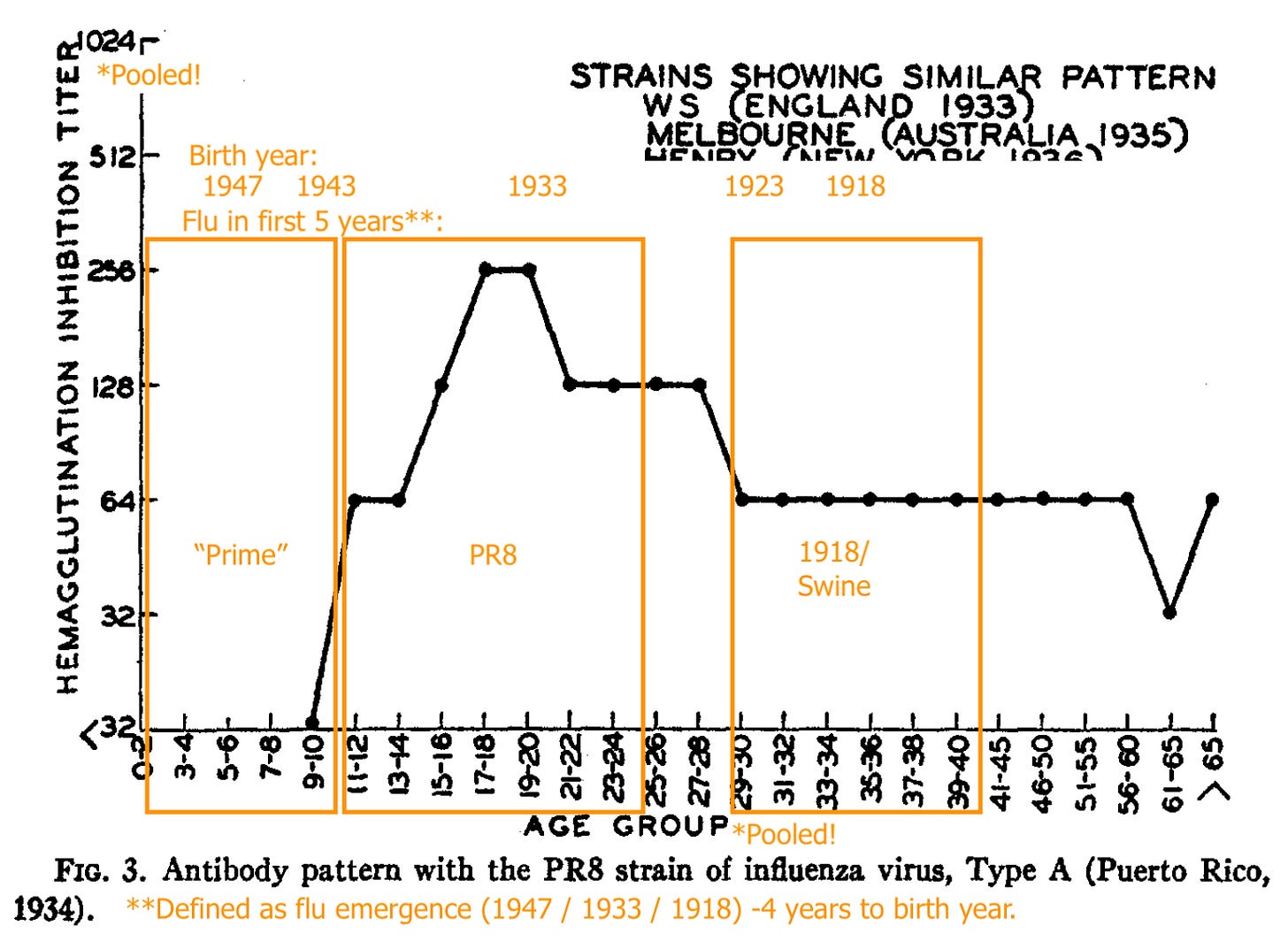
“Hood” (a representative of the post-1947 antigenic shift model) is only strongly recognized in the pooled samples of the young.

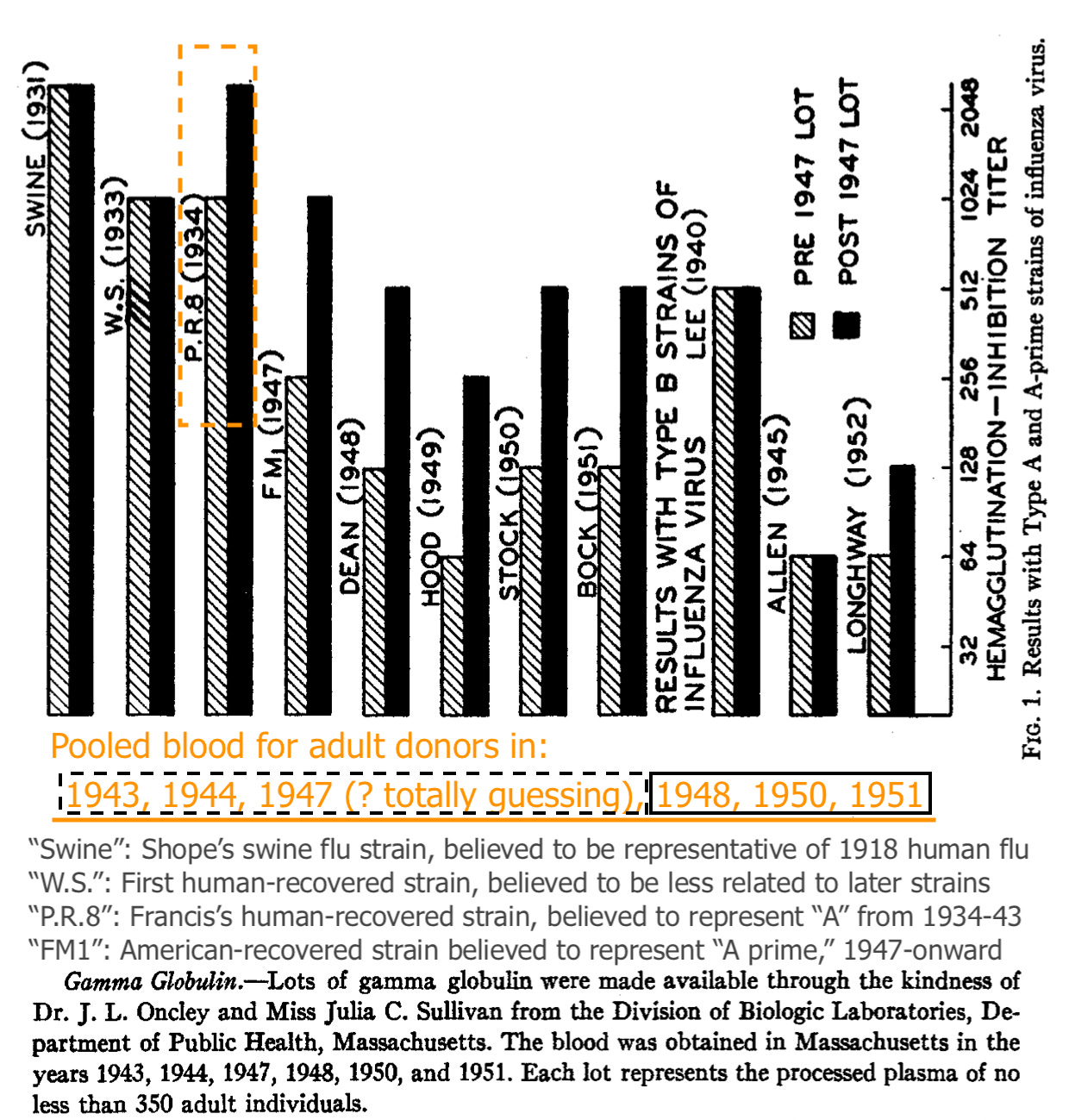



From this and the uptick in “pooled adult PR8 recognition” after 1947, Francis and co. infer that adults are failing to develop immune recognition against the newer lineage, and instead only increasing recognition against older strains.
This is a fallacy, as rates of infection are unknown, and low binding may simply reflect that not many adults have even encountered the virus in the first place.
Further, the stark contrast in HI patterns failed to be replicated in sera collected the same way the next year, especially when a 1947-vintage strain (“Rhodes”) was used to appraise A prime immunity:

The “orientation” of older adults toward the 1918 virus (represented by Shope’s swine flu) turned out to be an illusion.
3. How should “what Francis saw” be interpreted today?
The increase in recognition of PR8 after 1947 among “all adults” itself does not demand a particular interpretation of the by-age results. There is no “This could only be the case if, in these other ambiguous results, X obtains!” here. Rather, the results are intrinsically ambiguous:
Is it the case that 1918-immune adults were infected with PR8 as often as the next generation? If not, the failure of pooled blood to bind is a reflection of a high prevalence of non-PR8-infected.
Is it the case that many of them have been infected with “Prime”? If not, the failure of pooled binding is a reflection of a high prevalence of non-Prime-infected.
Is it the case that PR8-immune young adults have been infected with “Prime”? If not, the failure of pooled binding is (once again) a reflection of a high prevalence of non-Prime-infected.
All of these pooled results could reflect not lower rates of post-infection immune response, but lower rates of infection. Incredibly, the discussion section of the paper acknowledges this exact point as manifestly obvious (emphasis added):
With increasing age and repeated infections by antigenic variants of influenza virus, the population acquires a composite of antibody against a greater number of antigens. As a result, the serologic foundation of immunity is broader, and the incidence of influenza is lessened in the older age groups irrespective of the strain in circulation.
But an imaginary reader perusing the text backwards would do a double-take at the sensible statement above, as in the next paragraph, Francis and co. go on to claim:
The results of this study [scare-quotes wanted on “study”] also suggest that during the initial infections with influenza viruses, which occur predominantly in childhood, the major antigens of the prevailing strains have a unique effect upon the antibody-forming mechanisms which persists throughout life and largely determines the character of the future antibody with which this cohort of the population will respond when subsequently exposed to related strains of virus.
And so with this statement, “Original Antigenic Sin” makes its first bid for scientific legitimacy. But at which point, in the pooled serum results, was “subsequent exposure” demonstrated?
(It wasn’t.7)
Instead, Francis and co., with overt intellectual malice, are taking the post-1947 increase in pooled adult binding to demand of their reader a particular interpretation from their “no-resolution” by-age results (with a supplemental reference to outcomes in a failed “prime” vaccine trial), the same way an abusive child might coax a mentally disabled kid into action by convincing them they are in some type of big trouble. This is not the case. Regarding the pooled adult binding, it may simply be true that exposure to “prime” strains gave previously low-PR8-binders a boost in cross-reactive antibodies, leading to an increase in pooled binding; rather than that high-PR8-binders met “prime” and doubled down on PR8. And so the reader’s interpretation of the by-age results should not include any firm conclusion about whether “immune responses” or “infection rates” are driving the lower recognition of newer strains.
With such imprecise results as Francis and co. have offered, all the reader can really do is wildly guess.
With that said…
It is reasonable for the reader to wildly guess that exposure to a current strain elicits memory response against prior versions of flu - because that is how we understand immunity to work, and because such a response confers protection vs. no prior immunity.
It is reasonable for the reader to wildly guess that this occurred for adults infected with “prime” - but also, that if these adults were individually sampled, there would be clear evidence of a novel immune response against prime.
As an analogy, imagine that proficiency with iOS 7 is being examined in 2014. It would be demonstrable that the newest, youngest users of iPhones are most fluent with the newer features; and that the elderly as a group are not fluent at all.
Yet it would still be true that:
The elderly who used previous versions are able to use iOS 7 better than those who have yet to use iOS.
The elderly who encountered a difficulty with iOS 7 and contacted support, gained proficiency in the new feature they needed to learn. Otherwise, iOS 7 killed them.
You could combine the elderly into one set and obscure these truths; but they would still be true.
This is likewise how memory immunity works. It leverages prior knowledge and updates that knowledge when required. Anything else is a mathematical illusion.
4. Is “immune imprinting” any different from the current understanding of B Cell immunity?
The more intelligent literature concerning “OAS” tends to acknowledge the obvious balance described above, and yet still often portray it as some type of flaw. And so “antigen trapping” is used to describe the condition that prevails when, despite a variation in the flu virus, previous antibodies still work against the virus:
As others have speculated, it is plausible that the decreased antibody responses to subsequent exposures may be a result of “antigen trapping”, a hypothesis according to which binding of antigen by pre-existing cross-reactive antibodies and memory-cells decreases the antigenic load available for priming naïve B-cells and leads to a diminished novel response8
“Immune imprinting” might be even worse in terms of discussing an understood facet of the immune system - that immune memory is distributed through time, resulting in a tapestry of T and B Cell models that reflect the experience of the organism, like a yearbook or a quilt - and treat it as some great puzzle!
To pick once again on Röltgen, K. et al.:
The degree of imprinting may depend on the particular variants and the order in which they are introduced to the individual’s immune system, and the number of exposures, such as the number of vaccine doses received.9
What do they mean, “may”?!
What memory immunity must remember is that which it encounters.
If you derived value from this post, please drop a few coins in your fact-barista’s tip jar.


Potential changelog here.
The information in this portion is either general knowledge or discussed in:
Akkaya, M. Kwak, K. Pierce, SK. “B cell memory: building two walls of protection against pathogens.” Nature Reviews Immunology. volume 20, pages 229–238 (2020).
The mention (on the circuitry cartoon) of Germinal Centers continuing “if they want to” is further discussed in:
Yewdell, W. et al. “Temporal dynamics of persistent germinal centers and memory B cell differentiation following respiratory virus infection.” Cell Reports. Volume 37, Issue 6, 9 November 2021, 109961
Heesters, B. et al. “Endocytosis and Recycling of Immune Complexes by Follicular Dendritic Cells Enhances B Cell Antigen Binding and Activation.” Immunity. Volume 38, Issue 6, 27 June 2013, Pages 1164-1175
Lots of B Cell memory responses happen outside of germinal centers, as discussed in Akkaya et al. above and:
Chen, J. et al. “High-affinity, neutralizing antibodies to SARS-CoV-2 can be made without T follicular helper cells.” Sci Immunol. 2022 Feb 4;7(68):eabl5652.
However, the durability of these types of responses is not well understood.
For further details on Natural IgM antibody, see:
Panda, S. Ding, J. “Natural Antibodies Bridge Innate and Adaptive Immunity.” J Immunol January 1, 2015, 194 (1) 13-20;
Kim, D. et al. (2011.) “Insights into the regulatory mechanism controlling the inhibition of vaccine-induced seroconversion by maternal antibodies.” Blood. 2011 Jun 9;117(23):6143-51. doi: 10.1182/blood-2010-11-320317. Epub 2011 Feb 28.
Davenport, FM. Hennessy, AV. Francis, T Jr. “Epidemiologic And Immunologic Significance Of Age Distribution Of Antibody To Antigenic Variants Of Influenza Virus.” J Exp Med. 1953 Nov 30; 98(6): 641–656.
At best, one could argue that the phrase “this cohort” is meant to refer to responses in cohorts as pooled, not to responses among individuals within cohorts. Concluding the former would allow us to exonerate Francis from all subsequent interpretations of OAS that obviously take it to mean the latter. However, this would render as patently irrelevant all elements of the discussion which refer to infections despite prior exposure (as opposed to protection from infection), as in the second part of this quote (emphasis added): “These experiences confer a broader immunity which limits infection with, and antibody response to, the more recently encountered strains. The antibody-forming mechanisms appear to be oriented by the initial infections of childhood so that exposures later in life to antigenically related strains result in a progressive reinforcement of the primary antibody.”
Fonville, JM. et al. “Antibody landscapes after influenza virus infection or vaccination.” Science. 2014 Nov 21; 346(6212): 996–1000.
(As always, modern flu research is a creature of its obsession with yearly vaccines, and an unnatural existence free from the occasional hard knock to the lymph node: There can be no question of the immune system already having the optimal system for husbanding resources in reply to a constantly evolving virus. Anything it does to resist being forced around like putty is a “sin.” In this paper, however, the authors provide resounding evidence against the myth of OAS.)
Röltgen, K. et al. “Immune imprinting, breadth of variant recognition and germinal center response in human SARS-CoV-2 infection and vaccination.” Cell.
Previously reviewed in “Even-Steven.”
There is a new twist to the story in the case of those >/= 2X transfected with mRNA.. and that is IgG class switching toward "non-inflammatory" IgG4 antibodies. This is discussed in a new paper out of
Germany that I feel has gotten too little attention by those oriented toward immunology;
https://www.medrxiv.org/content/10.1101/2022.07.05.22277189v1.full.pdf
IgG4 antibodies no longer fight the infection - rather they are a sign that the immune system is tapping out due to overstimulation by an antigen. "Vaccines" are not supposed to do this. The mRNA is clearly hanging around way too long..................
I was a little concerned when you actually prefaced this post with a "nitty & gritty" remark. I thought we were going to be taken through an entire year of medical school!
I ended up looking up Frances' OAS argument, which appeared in early 1960s (possibly 1960 itself? Although your post of 1953 likely aligns with his initial research timeline). Much of the B-cell research appear to have happened a few years after, with many research coming about during the late 1960s to the 1990s (and onward, really) if the timeline from this article is to be believed.
https://www.nature.com/articles/nri3801
And so that would really line up with the notion that antibodies and OAS were decided before much was known about B and T cells as we do today, and why there are many issues with what we are having right now.
I wonder if much of the talk of OAS has overridden people's conceptions about the adaptive immunity. It is, in essence, a requirement that the adaptive immune system rely on recall in order to tackle a pathogen that shares some similarities to prior ones. So now we have prior immunity recalling a response, and it's assumed to be immediately bad. I even remember reading a part of one of the first mouse studies on OAS where the researchers even commented that the recall would have been a good thing.
I also guess this is all a consequence of everyone just focusing on antibodies and nothing else. The whole immune system is complex and can't be boiled down to the antibodies sticking once, and if they continue to stick again.
I look forward to the Frances portion and see what you write about that!