Severe “Covid-19” in turn causes adaptive immune suppression. (go)
Other examples of T Cell suppression and immune “stalemate” in persistent disease. (go)
The likely implications of IgG4 tolerance in light of the reality of severe “Covid-19”: persistent infections, ADE? (go)
Part 2: So just what is “Covid-19 pneumonia”? While much attention has been diverted to the interplay of the virus and spike protein on the vasculature, it still seems unusual that the etiology of disease in the lungs should remain so vaguely understood and described.
i. The Brian Mowrey Theory of Covid-19 Pneumonia
Whatever the merits of the case I have waged that “cytokine storm” was a front-loaded myth that never materialized and remains refuted by all the evidence, it is nonetheless true that the expert consensus is still that severe disease is an immune-mediated malady. First, virus suppresses the antiviral response. Then, the innate immune system realizes it “missed the bus” and “goes berserk.”
This consensus, I argue, ignores — or simply waves away — all the evidence of what the virus actually does to the lungs.
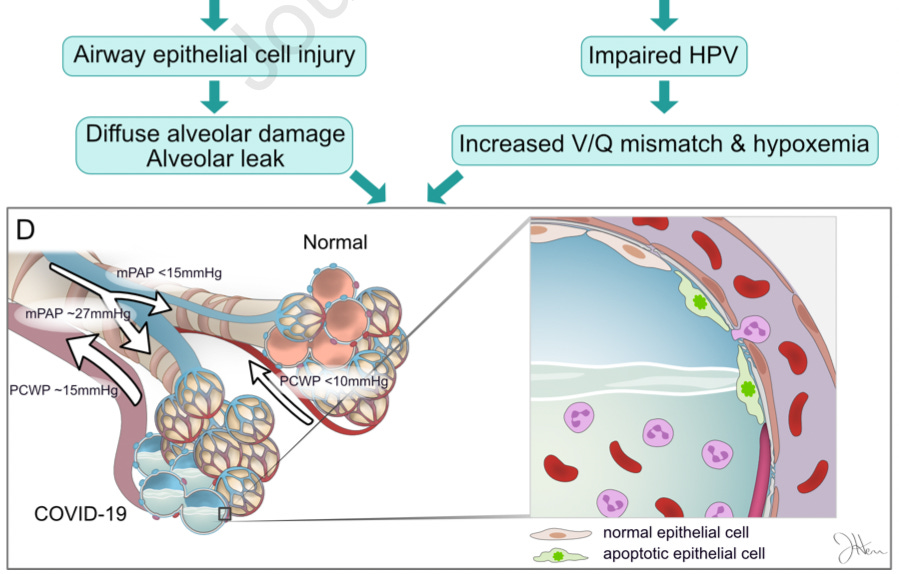
1. What the virus actually does
Here my thinking is heavily influenced by Heiss, et al. and Archer, et al., which are jointly discussed in the “Air-less lung areas” post:
Archer, et al. propose that the virus shuts down the reflex that normally prevents blood flow from going to lung regions that have obstructed airflow (hypoxic pulmonary vasoconstriction, or HPV).1 One could imagine little air-sacs as water-logged engines. Normally the body would shunt blood away from these flooded zones; failure to do so is like trying to turn the engines over in water.
From lead author’s Stephen L Archer’s The Conversation summary:2
People with severe COVID-19 pneumonia often arrive at the hospital with unusually low oxygen levels. They have two unusual features distinct from patients with other types of pneumonia:
First, they suffer widespread injury to their lower airway (the alveoli, which is where oxygen is taken up).
Second, they shunt blood to unventilated areas of the lung, which is called ventilation-perfusion mismatch. This means blood is going to parts of the lung where it won’t get sufficiently oxygenated.
Together, these abnormalities lower blood oxygen.
After describing the reflex that keeps blood from going to airless parts of the lung (which the authors postulate is regulated by mitochondria, though this is a bit of a pet theory on their part):
This mechanism has an important function. It directs blood away from areas of pneumonia to better ventilated lobes of the lung, which optimizes oxygen-uptake. By damaging the mitochondria in the smooth muscle cells of the pulmonary artery, the virus allows blood flow to continue into areas of pneumonia, which also lowers oxygen levels. […]
The results of [mitochondrial] damage — an increase in apoptosis in airway epithelial cells [this is possibly an innate immune defense mechanism, as infected cells self-destruct], and loss of hypoxic pulmonary vasoconstriction — were making lung injury and hypoxemia (low blood oxygen) worse.
To my mind, this is sufficient to explain the observed injury, without any need to implicate immune over-reaction. Maybe I’m just crazy, but it sounds like the virus is perfectly capable of destroying lots of lung cells on its own, unchecked, thanks to suppression of antiviral innate immunity.
2. Organizing pneumonia suggests generic injury
“Organizing pneumonia” is an all-but-necessary sequelae of this destruction:3
Several reports have now provided radiological evidence for different forms of diffuse parenchymal lung disease (DPLD) during the later course of the disease.
Organising pneumonia (OP), a form a DPLD, is a distinct clinicopathological entity that may occur as a pulmonary reaction to various injuries, including ARDS.
OP is characterised by the patchy filling of alveoli and bronchioles by loose plugs of connective tissue with concomitant diffuse alveolar damage, the hallmark of ARDS. Radiological findings typically include peripheral consolidation, ground-glass infiltrates and/or solitary nodules. The definitive diagnosis of OP requires histological assessment and the primary treatment of OP [in general] is, apart from treating the underlying disease, corticosteroid administration over several months, initially at relatively high doses.
“Covid-19 pneumonia” ultimately results in acute lung conditions and exam findings that, depending on the period of disease, are consistent with:4
pulmonary edema [fluid in lungs], organizing pneumonia, or lung injury of any other cause (e.g., drug toxicity, radiation treatment, or a cryptogenic cause), pulmonary infarcts [pulmonary embolism], alveolar hemorrhage, and interstitial lung diseases (e.g., nonspecific interstitial pneumonia or desquamative interstitial pneumonia)
None of this argues strongly for autoimmune destruction.
Archer, et al. provide evidence that multiple viral proteins alone may trigger cell death responses from mitochondria, suggesting that infected cells could operate as toxin factories; this may explain the resemblance in CT scans to drug and radiation injury.5
And note the unsurprising suggestion that the virus is also indirectly injuring the lung via clotting (infarcts). It is well established that lung pathologies in severe Covid-19 are partly vascular, from micro-clotting (thrombotic angiopathy) to full-on pulmonary embolism.6 In autopsies of 12 consecutive patients dying after PCR-confirmed infection with SARS-CoV-2:7
Autopsy revealed deep venous thrombosis in 7 of 12 patients (58%) in whom venous thromboembolism was not suspected before death; pulmonary embolism was the direct cause of death in 4 patients. Postmortem computed tomography revealed reticular infiltration of the lungs with severe bilateral, dense consolidation, whereas histomorphologically diffuse alveolar damage was seen in 8 [67%] patients. In all patients, SARS–CoV-2 RNA was detected in the lung at high concentrations
Tissue that is deprived of blood dies (and sometimes the body with it), without any need for the immune system to step in and "over-react" to the lack of blood.
The patients in the Wichmann, et al. autopsy series did not develop pulmonary embolisms and Diffuse Alveolar Damage after the virus left the scene; it was still active in the lungs in all cases.
This does not support the still-reigning characterization of Covid-19 pneumonia as a secondary immune attack following a bit of harmless virus-cell hookup.
ii. Looking at what “works”
The etiology of Covid-19 pneumonia ought to be consistent with treatment outcomes. If X is really the cause of the problem, then targeting X will demonstrably prevent the problem. I submit that the evidence here argues for the virus as the direct agent of lung tissue destruction, not the immune system’s response to it.
Here, we will examine five “interventions” and their effects on severe outcomes: Changing the virus; steroids; early seroconversion, Paxlovid, and (trigger warning) the experimental Covid vaccines.
1. Changing the virus
Notably, when consecutive autopsies of death after infection with the Omicron siblings (BA.1 or 2) were performed, results were starkly different:8
Fatal cases after Omicron BA.1 and BA.2 infection: Diffuse alveolar damage occurs only in a minority
Despite high viral loads in almost all nasopharyngeal swabs and in 13 lung tissue samples, death caused by COVID-19-associated diffuse alveolar damage (DAD) in the acute and organizing stages was found in only eight cases (31%). This rate is significantly lower compared to previous studies, including non-Omicron variants, where rates of 92% and 69% for non-vaccinated and fully vaccinated vaccines were observed [note that these prior results were from two separate, non-simultaneous autopsy series involving different variants as well].
Did the human immune system start functioning differently overnight when the Omicrons displaced ancestral strains, or the virus (it was the virus)?
2. The limits of immunomodulation
Steroid-based therapies for Covid-19-induced organizing pneumonia are not different from steroid-based therapies for any other type of post-injury organizing pneumonia. To repeat the previous excerpt, “the primary treatment of OP is, apart from treating the underlying disease, corticosteroid administration over several months.” Modulating inflammation can be beneficial after any type of injury. This does not mean the injury was caused by inflammation.
Moreover, if an innate immune “over-reaction” were causing Diffuse Alveolar Damage to begin with, one would expect more impressive outcomes from steroid treatment during acute disease.
Here, our conclusions are limited by the question of whether steroid treatment after hospitalization is also “too little, too late,” as likely true for other therapies like hydroxychloroquine which performed poorly in inpatient settings. Nonetheless, results seem unimpressive; offering no support for the consensus theory that immune over-reaction causes lung damage to begin with.
Thus, steroids can ameliorate Covid-19 pneumonia — and due to suppression of antiviral immunity may even cause harm if given too early (in theory, and consistent with the RECOVERY results; with the caveat that some smaller studies have found better outcomes).
Can results from administering steroids so late in the disease course really speak to the cause of disease? In theory, inflammation-mediated tissue damage would still be ongoing after hospitalization for most or at least some patients — hence why I highlight the different background death rates in the two studies above, which should show a difference from earlier intervention.
Overall, there is almost certainly an important role for steroids in modulating inflammation at some point in the course of and after severe Covid-19. Just as after any injury.
But they do not appear capable of preventing the core injury at all; thus, they do not support the theory that the injury is caused by immune “over-reaction.”
3. Early seroconversion protects
The Paxlovid trial achieved what no other study has apparently managed to do (cleanly12) to date, which is discern how to predict severe outcomes in an at-risk population who have recently tested positive for the virus.
Among the placebo group, roughly half of placebo participants happened to already be positive for antibodies (IgG or IgM) at the time of randomization. 58 of 66 severe outcomes occurred in the antibody-negative placebo participants.
Thus, (at-risk!) people who had an early antibody response were highly protected. Their immune system had detected the virus promptly; it was “on the job,” and viral tissue destruction was limited. (See “Paxlovid Trial Curiosities” for full discussion.)
This strongly supports the theory that severe outcomes result from uncontrolled viral replication due to suppressed antiviral immunity. Granted, the consensus view also acknowledges suppressed antiviral immunity and viral replication preceding the supposed “over-reaction” to tissue destruction.
However, once one considers the direct evidence that a slow immune response predicts likelihood of severe outcomes better than any other sub-factor in the at-risk, it becomes clear that the immune “over-reaction” is a totally unnecessary part added on to disparities in direct viral destruction. I.e., the “over-reaction” theory is somehow proposing that there is a threshold of destruction that must be reached for this “over-reaction” to… cause the destruction to begin with.
4. Paxlovid protects as well
Love it or hate it, Paxlovid kept at-risk, unvaccinated trial participants from developing severe outcomes just as effectively as early antibody production did in the placebo group.
How did it achieve this miracle? By halting viral replication. While real-world results continue to support severe efficacy (even in the vaccinated), they are more subject to potential biases.13 However, the notorious rebound phenomenon itself offers direct proof that Paxlovid is halting viral replication (because things cannot restart that have not been stopped). (See “Unfinished Business” for more on the biomechanics of rebound.) Once the virus gets back to work, after 5 days of suppression, it is no longer “outrunning” the slow-footed immune response among the at-risk — they too now have antibodies to help limit viral replication and tissue destruction.
5. Experimental gene transfection injection protects as well
Despite starting the Price-Is-Right Cliffhanger march toward tolerance, the Covid vaccines protect against severe outcomes (at least for the short term).
ACTUALLY COULD I CHANGE MY BID, BOB
How do they achieve this miracle? By halting viral replication.
The likely mechanism is via IgG transudation (seeping) into the lungs, especially the lower lungs. IgG antibodies can also protect the lungs via tagging infected cells and virions for destruction by innate immune cells, preventing the problem that leads to severe outcomes to begin with (i.e., slow immune responses).
As an analogy, imagine the Covid vaccines as setting up a fire station in the woods. The firefighters can still help when it turns out the arsonist prefers suburban targets. This will not prevent the fires from being started (i.e., there is no infection immunity except when serum antibodies are so high that the respiratory tract is flooded with them), but it will help limit destruction. There is plenty of precedent with other viruses. For RSV (a virus in which the case for immune-mediated pathology is rather stronger, notably), it is clear that IgG is protective of severe outcomes, especially in the lower respiratory tract:14
Virus-neutralizing antibodies in the respiratory tract likely contribute to viral clearance and certainly play an important role in protection against reinfection. They include secretory immunoglobulin A (IgA) and transudated, serum-derived IgG. […] Serum IgG antibodies are somewhat more efficient in accessing the lower respiratory tract and can provide substantial protection in that compartment. In RSV-naïve infants, the maternal serum antibody titer is positively correlated with a reduced level of severe RSV disease. The clinical experience with palivizumab also shows that serum antibodies alone can provide substantial protection [55% in at-risk infants] from severe disease.
Conclusion: The keep-it-simple model will prevail
We thus have 3 examples where stopping the virus alone protects against Covid-19 pneumonia (and other severe outcomes), and no apparent examples of such a dramatic effect from immune modulation alone. And, we have an example of changes to the virus obviating pneumonia, which does not support the immune “over-reaction” theory.
I believe that in the long run, Archer, et al.’s findings will be recognized for their importance; the unnecessarily complicated consensus of immune-mediated tissue damage will become passé; and a “keep it simple, stupid” model of the disease will prevail. For the moment I am calling this model “The Brian Mowrey theory” just to emphasize how out-of-touch this model is with the consensus.
In Part 3: Oh yeah, but this was supposed to be about tolerance wasn’t it?
These effects may be common to multiple coronaviruses; as partially confirmed in Archer, et al.’s own experiments. However, we typically encounter these viruses in childhood when the innate immune response is at its most nimble and effective.
Plenty of studies have shown associations with lack of at-randomization seroconversion and severe outcomes; but these did not involve standardized times-from-symptom-onset and thus could not be meaningfully interpreted in terms of “early” vs. late seroconversion. The Paxlovid trial placebo group’s “natural immunity assay” introduces a standard time-from-symptom window (0-5 days, with a skew toward 0-3 days, 735 of 1,126 placebo recipients) and probably defaulted to milder early disease states due to the need for active trial enrollment.
-Note <20 week since last dose group will have artifacts of changing age groups since two waves were observed, and the “second booster” was authorized in the first wave.
A third study has now made a big splash on this topic; however, this one is less impressive due to the “control” group including so many same-day hospitalizations (which Dryden-Peterson took care to exclude).
I'm pretty sure there a quite a few ivermectin users who would agree with the Brian theory.
After my daughter's 2nd covid (Oct 2020) her legs no longer worked and she became incontinent....among other horrid symptoms like intense pain. No doctors would treat her for this apparently psychiatric illness....yeah
Ok. So after 8 months of being bedridden, with the help of a kind soul, we found ivermectin. My daughter was walking again in two days.
I don't believe cytokine storm, immune dysfunction, or psychosis can explain that.
I do believe if we'd had ivermectin much sooner than over a year after 1st infection and 8 months after 2nd infection, that she wouldn't be a long hauler. It was too little too late, but still a miracle.
As it was her only med, it must have acted directly on the virus.
If people could speak of ivermectin...or actually USE it, there'd be that much greater understanding of the virus. But the censors/overlords can't have that happening.
Why use a safe med like ivermectin when you can kill and maim with a shot?
I remember John Paul wrote an interesting sub stack on bacteria binding to subunits of the spike protein.
"the Spike Protein interacted with (bacterial) LPS to form aggregates of Spike Protein",
"While using different pathways, a lot of the recent deaths directly from vaccination (as per the pathologists analysis) and from the infection since early 2020 have something in common. Bacterial overgrowth, excessive presence of bacteria in the blood."
Thanks for the trigger warning! With so much concern about long-term side effects of these injections, I've also kept in mind that they seem to have a short-term benefit on the very low risk of severe infection.













I'm pretty sure there a quite a few ivermectin users who would agree with the Brian theory.
After my daughter's 2nd covid (Oct 2020) her legs no longer worked and she became incontinent....among other horrid symptoms like intense pain. No doctors would treat her for this apparently psychiatric illness....yeah
Ok. So after 8 months of being bedridden, with the help of a kind soul, we found ivermectin. My daughter was walking again in two days.
I don't believe cytokine storm, immune dysfunction, or psychosis can explain that.
I do believe if we'd had ivermectin much sooner than over a year after 1st infection and 8 months after 2nd infection, that she wouldn't be a long hauler. It was too little too late, but still a miracle.
As it was her only med, it must have acted directly on the virus.
If people could speak of ivermectin...or actually USE it, there'd be that much greater understanding of the virus. But the censors/overlords can't have that happening.
Why use a safe med like ivermectin when you can kill and maim with a shot?
I remember John Paul wrote an interesting sub stack on bacteria binding to subunits of the spike protein.
"the Spike Protein interacted with (bacterial) LPS to form aggregates of Spike Protein",
"While using different pathways, a lot of the recent deaths directly from vaccination (as per the pathologists analysis) and from the infection since early 2020 have something in common. Bacterial overgrowth, excessive presence of bacteria in the blood."
https://hiddencomplexity.substack.com/p/spike-protein-as-a-endotoxin-delivery
Thanks for the trigger warning! With so much concern about long-term side effects of these injections, I've also kept in mind that they seem to have a short-term benefit on the very low risk of severe infection.