Over at his substack, Igor Chudov highlights an apparent trend of online anecdotes: Patients treated with Paxlovid report a regression of symptoms and test-positivity. Here, I volunteer my off-the-cuff theory of why this could plausibly be “A Thing.”
The following theory was written before stronger evidence emerged that supports both the rebound trend and my explanation for it, which results in this series having a certain meandering structure. A guide:
“Unfinished Business” Table of Contents
Part 1 (this post):
Introduction: Early anecdotes
Official Unglossed Totally Reckless Theory Outline
Theory Explanation
Considerations for Covid-vaccine or Omicron interactions
Chudov collects 7 anecdotes (minus 2 in which treatment or test positivity are unclear); almost all report completing the normal 5 day course of Paxlovid:
“Annie,” 3rd-Dosed in December.
March 26: Antigen test+, symptoms+, Paxlovid started
April 2: Antigen test-, symptoms-
April 11: Antigen test+, symptoms+
“Feisty-Implement5642,” 3rd-Dosed in December.
March 10: Test+, symptoms+, Paxlovid started
March 12-15: Antigen tests- 4x, symptoms-
March 18: Test+, symptoms+ and worsened
“Haskell-Tres”
-10 Days from post: Test+ (faint), symptoms+ (mild), Paxlovid started
-3 Days: Antigen test-, symptoms-
Day 0: Tests+ 2x (stronger line), symptoms-
Husband of “real415”
End of March: Test+ (faint), symptoms+, Paxlovid started
Neutropenia (low-neutrophil-count)-having friend of “Barbara,” 3rd-Dosed
February 12: Test+, symptoms-, Paxlovid started
February 21: Test+
“John”
-2 Weeks: “Covid”+, Paxlovid started
-1 Week: Antigen test-, symptoms-
Day 0: Test+, symptoms+
Eric “#CovidIsNotOver” Feigl-Ding
Day 0: Test+, symptoms+, Paxlovid started
Day 6: Test+, symptoms+ (“5%” of before)
No need at this point for the caveats that were included in the initial version of this post.1 The above early anecdotes were indeed part of a broader trend, with later evidence corroborating the theory offered in this post to account for the trend. However, the designation “Totally Reckless Theory” given in advance of that later corroboration is retained for consistency throughout the series.
And so, presenting:
The Official Unglossed Totally Reckless Paxlovid Viral Reservoir Theory!
Paxlovid’s binding to the nsp5 / 3C-Like protease merely “pauses” the virus's work within already-infected cells.
This “pause” is pinned to the moment after the polyprotein is formed, but before nsp5 has chopped it up into little parts (including more nsp5's).
Pausing replication here means that the difference between clearance and regression is just a matter of which has a longer half-life in the body/cells: The drug, or the RNA and un-processed polyproteins of the virus?
There will be instances when the “pause” is final or illusory. If the payload of Paxlovid in the cell is enough to inhibit all nsp5s in every polyprotein, it will obviously be final - the polyproteins will be uncleaved. If the payload only partially inhibits nsp5, the pause will be illusory. This is due to the auto-cleaving nature of nsp5, and to the different efficiencies of action of uncleaved and cleaved nsp5. As the “not-Pax’d” nsp5s become cleaved (by each other), they become more efficient at cleaving (each other and the rest of the polyprotein). Without Paxlovid, this leads to a critical mass where cleaved nsp5s are abundant enough to “combust” the remaining nsp5 fuel and the other cleavage motifs of the polyprotein(s) in turn. With Paxlovid, this critical mass merely takes longer to arrive at (simplistic example: if 20% of nsp5s are “Pax’d,” then critical mass requires 25% more nsp5 to be cleaved before combustion. At this point, as above, replication resumes if the RNA has survived.
So unless the drug is taken for longer (until all the RNA is broken down) or forever, there may be some chance of viral regression from leftover polyproteins; the only question is “how much.”
Chesterton’s Protease
For correction notice on this segment, see footnotes.2
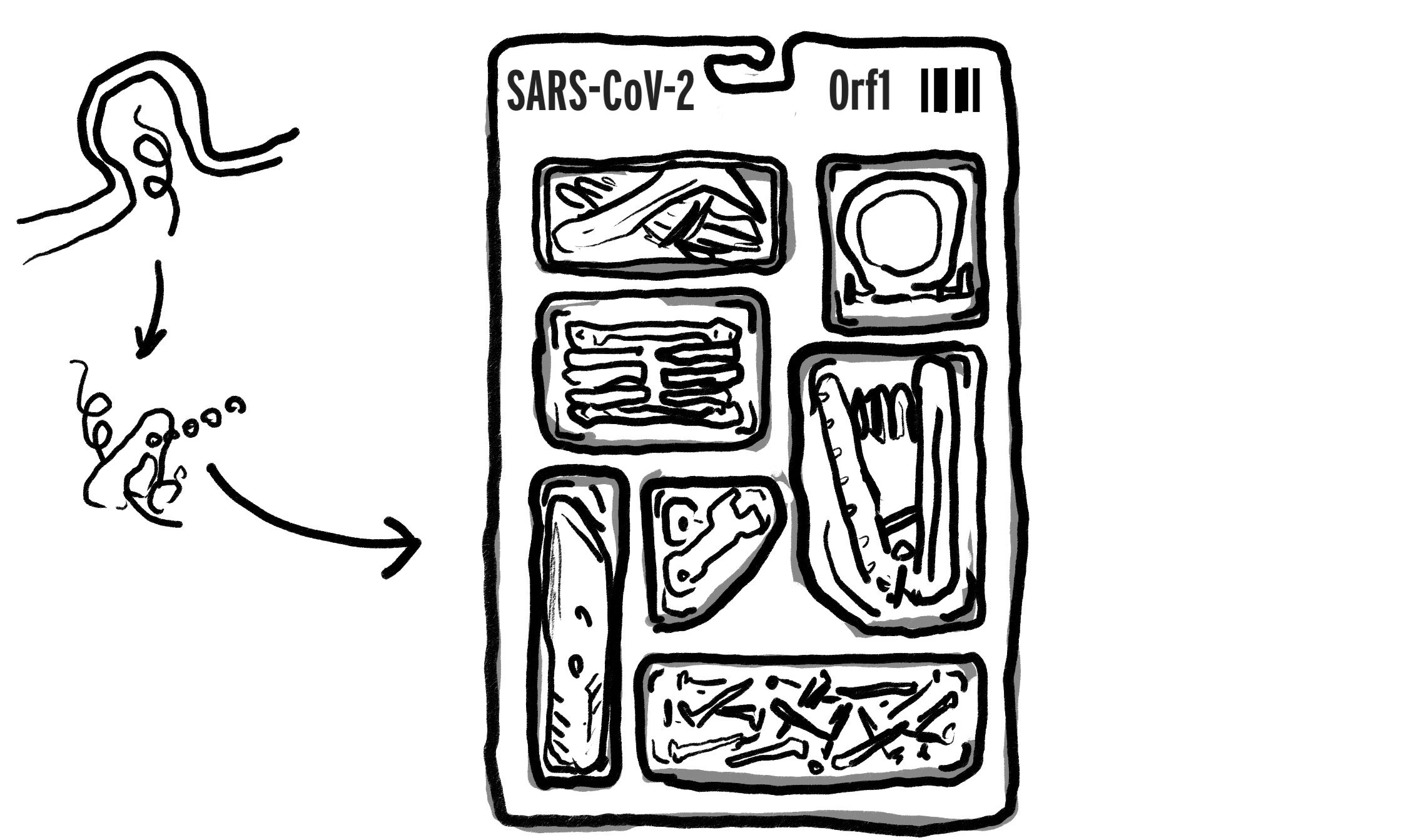
Here is how Paxlovid’s primary mode of action fits into the SARS-CoV-2 replication cycle:
When the virus’s RNA first enters the cell, it is read by a ribosome in the cell in order to produce loads of copies of the Orf1 “polyprotein,” which can be thought of as a single package containing a bundle of smaller tools that are useful for constructing more copies of the virus (it’s actually two packages, but that’s not important here, since both require nsp5 to an extent; anyway imagine the tools are split between two packages in the image if you want):
Some of the tools are responsible for sabotaging the host cell immune response; some for making new negative and positive copies of the RNA molecule (or proofreading and structurally modifying the same); and two, including that box-cutter on the lower left, for removing the other tools from the package.
Said box-cutter protein is called “nsp5” (non-structural protein #5), or the 3C-Like Protease, or “MPro.” (I am using the unconventionally generic “nsp5” designation because I think it contributes to more flexible thinking about the problem, vs. the logic that “the main coronavirus protease” is intrinsically a single target that can be knocked out via molecular sniper-shot.) Because the “blade” of this box-cutter uses a Cysteine residue to break apart links in the polyprotein, it is also called a “Cysteine protease” (or proteinase). So that’s the story behind the protein with four names that is targeted by the drug with three names (I am using “Paxlovid” because further distinction is superfluous).
Of course, the placement of the box-cutter inside the package that needs to be cut open raises an obvious chicken-and-egg-esque question: Exactly how does SARS-CoV-2 get this tool out of the same package that requires said tool to open it?
It may be the case that proteases which target the same “chopping motifs,” at least weakly, are already produced by human cells in a low rate; however the evidence is stronger that nsp5 cleaves itself. Imagine two copies of the toolset above pressing against each-other until the blade cuts through the plastic around the box-cutter, liberating it.
And once that happens, the box-cutter is suddenly much better at cutting out other box-cutters. And those become much better at doing the same. And so cleavage gets faster, and faster, etc., in a manner akin to combustion. Like a pile of charcoal coming to light after a few minutes of smoldering.
So this where the “protease inhibitor” molecule in Paxlovid ostensibly comes in: It binds to the “blade” of nsp5, so that it cannot cleave. Result: Virus doesn’t replicate. Or so it would seem.
For now, bear in mind that in any scenario where Paxlovid “works” - i.e., stops replication - infected cells will have some number (probably a large number) of unopened polyproteins, with not enough functional (“non-Pax’d,” liberated) nsp5’s to process them, as well as the original SARS-CoV-2 RNA molecule, and a bunch of free-floating proteins from the smaller, leader portion of Orf1 that are cleaved by a different protease which was not targeted by the drug. This illustration corresponds to the first version of this post and needs to be modified (again, see correction notice in footnotes):
The key flaw is that this “arrest” of progress takes place before nsp5 has been fully, or even substantially released from the polyprotein. Either Paxlovid prefers binding released nsp5s, in which case it is like a trap ensnaring the box-cutter right at the moment of “becomes faster at cutting,” or it binds without preference, in which case a given percentage of nsp5s both don’t help to liberate other copies when packages are “rubbed together,” and are of no use if liberated by a non-Pax’d copy.
Either model leads to eventual recovery of replication. We can imagine a scenario involving a gang of lockpickers stuck in a room, the door of which must be picked once for each lockpicker to escape, but with all already-liberated lockpickers able to assist in efforts from the other side. Normally, this means that escape from the room is very slow at first, less slow as some lockpickers make it to the other side, and then, at a certain critical mass of liberated lockpickers, becomes functionally instantaneous.
Normal (No Paxlovid) Model
“I can’t believe it’s not MS Paint!”
Escape slow at first. Since cleaved nsp5 becomes more efficient at cleaving other nsp5s, every liberation accelerates further liberation. “Time” is arbitrary in all frames, and merely illustrates “slow” vs “fast.”
Accumulation of more lockpickers took less time than the “first liberation.” At a certain local concentration of liberated nsp5, future cleavage of polyprotein-bound nsp5 becomes relatively instantaneous.
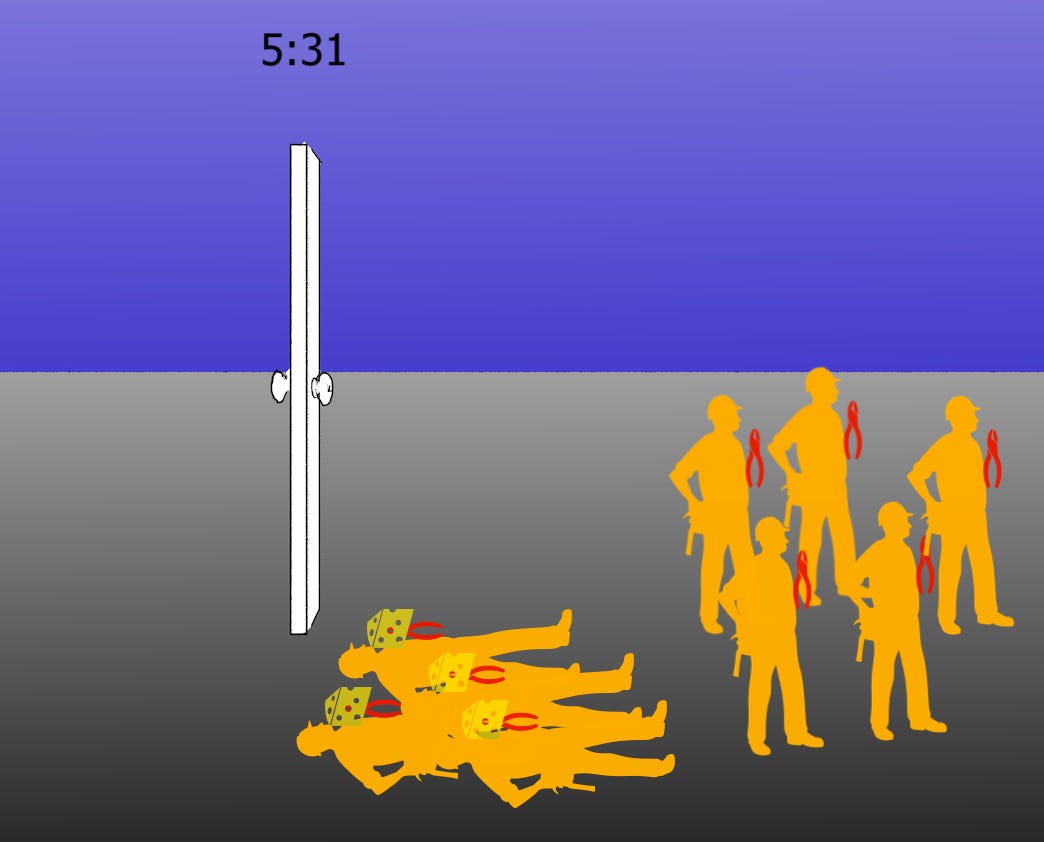
Off they go, to cleave the rest of the Orf1 polyprotein! Viral replication can now take place in earnest.
Paxlovid Model 1: Preferential
In the preferential model, Paxlovid is an opposing gang of lockpicker-huggers on the free side of the door. As long as there are more lockpickers than lockpicker-huggers, the lockpickers will win; particularly after the treatment course ends but also perhaps during treatment (notably, dosage was attenuated during the drug’s development). All the lockpickers have to do is keep coming out until there are no more free huggers to hug them; at this point, escape becomes faster and faster with each escape, etc.
“Preferential” Paxlovid binding model: Paxlovid prefers to bind cleaved nsp5
Liberated nsp5 preferentially bound by Paxlovid. Liberated nsp5’s do not contribute to further liberation. (No acceleration, in fact likely marginal deceleration.)
After a stiff initial penalty, liberation of nsp5 at last begins to accelerate, resuming the normal progression to critical mass. Paxlovid-bound nsp5 is basically junk protein, so let’s just knock those ones over. Further penalty for lower action of bound nsp5 at this poiint vs frame 2 of the normal process is not calculated.
Off they go! Can’t keep a good nonstructural protein #5 down.
Paxlovid Model 2: Non-Preferential
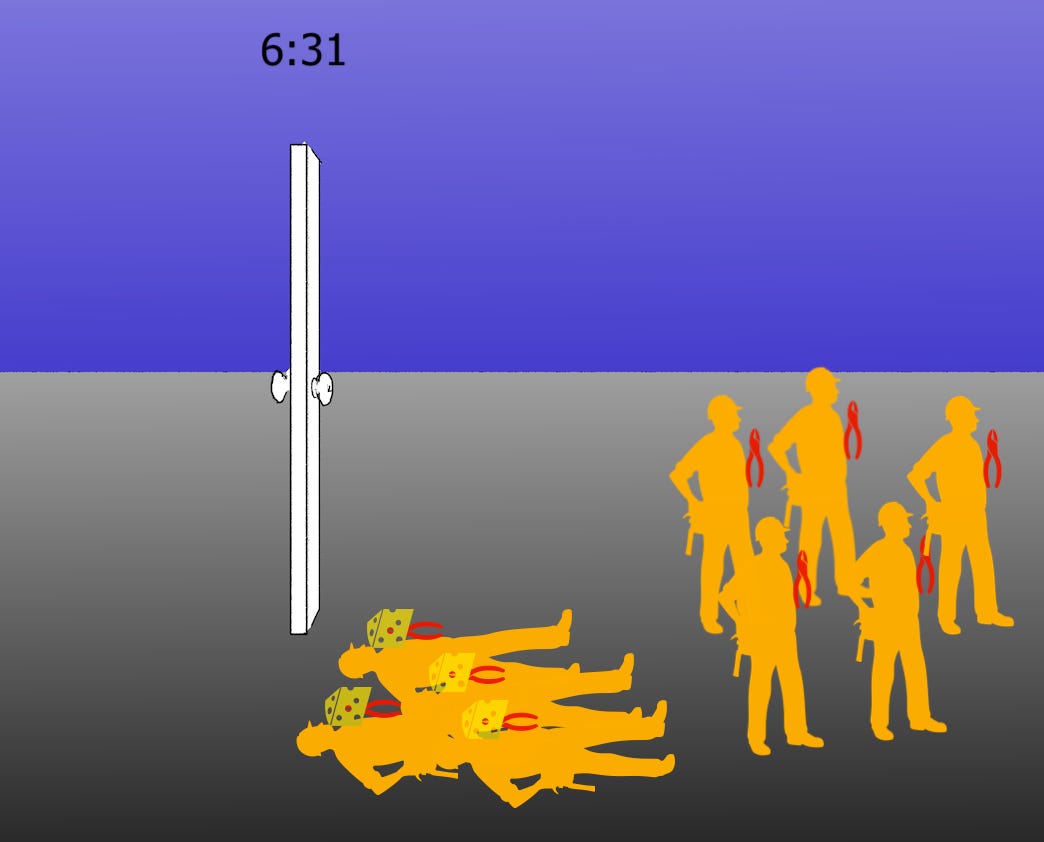
In the (purest form of the) nonpreferential model, the lockpicker huggers hug the lockpickers while they are all still in the room; but as before there may be fewer huggers than lockpickers. But here the metaphor is not perfect, because there is not actually one door and one lockpicker at a time trying to work it; so, scrap the door part. Just keep in mind that there is a room, and an outside, and some number of lockpickers have been hugged. The hugged lockpickers do not help when they are inside the room, nor if they are let out of the room by the non-hugged lockpickers. But the non-hugged lockpickers still help. So every non-hugged lockpicker that gets out of the room increases the rate of freeing all lockpickers. Eventually enough are outside of the room that freeing the whole lot takes no time at all (exactly as happens when there are no huggers to begin with).
“(Pure) Non-Preferential” Paxlovid binding model: Paxlovid binds to nsp5 pre-cleavage
Things seem rough from the start. Even the imaginary “first cleavage” will be slowed.
Imaginary first cleavage takes an extra “1 fake time units.” However, now liberated, non-cheesed nsp5 can accelerate future liberation. Note that a cheesed nsp5 could be liberated first; but in this simulation non-cheesed were set to outnumber cheesed. Who knows what the normal ratio is.
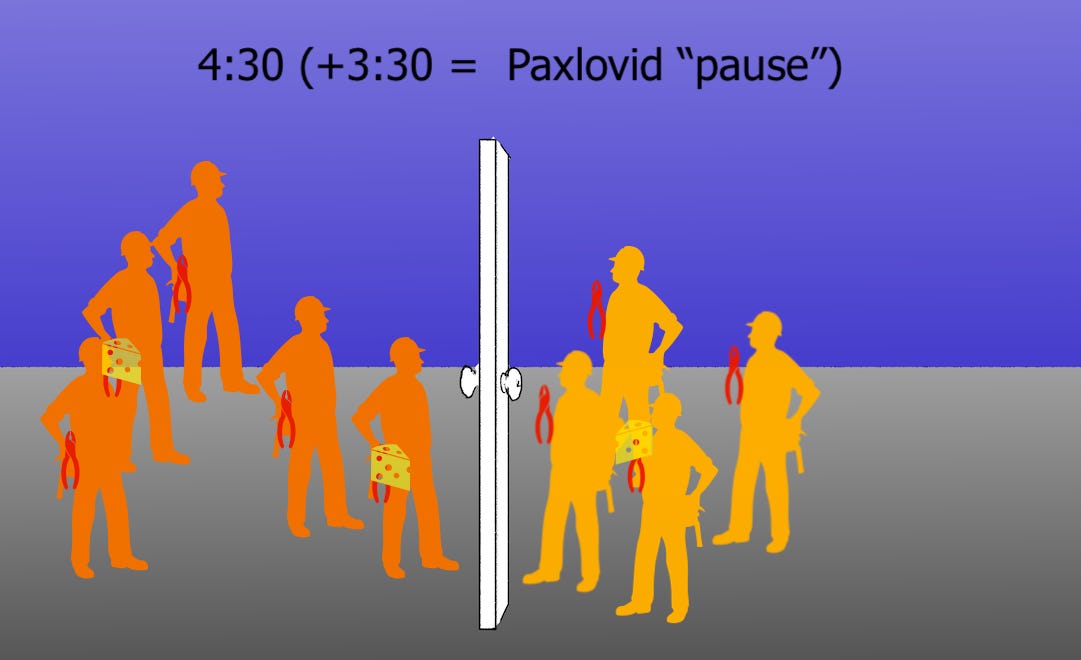
Paxlovid-bound nsp5 is still occasionally cleaved. These do not accelerate future cleavage. It is the non-cheesed nsp5s that are doing the cleaving, whether liberated or not. Thus, critical mass still requires more overall cleavage. We aren’t ready to count our effective “Paxlovid pause” time penalty until then.
There we go. “Critical mass.” (“Pause” should equal +2:30, oops.)
Back to work. (The clock should read 4:31, oops).
In either model, or any given blend, following the arrival of “critical mass,” the un-Pax’d lockpickers go around and open the doors of all the other rooms to liberate other members of the heist squad. The Orf1 polyprotein(s) becomes fully unpacked. Paxlovid only made it look like replication had stopped. In my arbitrary / intuition-based construction above, preferential is actually slower than non-preferential, despite appearing faster at first.
So, what’s to stop the virus from simply resuming production when this is achieved, either gradually during treatment or after the 5-day course of Paxlovid is complete? Namely, either:
All of the viral RNA in every cell must degrade by then, so that it does not matter if unopened Orf1 polyproteins are opened later.
Failing that, the immune system must locate and destroy every cell that has this assortment of “paused” viral replication elements within.
For both of these guardrails to fail, however, is robustly plausible: Where there’s a Nature, there’s a find-a-way. Even a 1% survival rate for viral RNA after a 5-day course would only set the virus back by about a day’s-worth of replicative progress. And if the innate immune system was not able to keep the virus in check in the first place, then there is no a priori reason to assume that it will be able to snipe all of the “paused” infected cells before the nsp5 box-cutters are released.
It Gets Better (Not)
Failure to take out infected cells before replication resumes is not just plausible in an abstract, “It could happen sometimes” sense; in fact, the virus has already leaned the odds in its favor. Because of course it has, because it’s not such an idiot to put all its eggs in one molecular basket (a lesson we humans seem to require learning again and again).
Here, again, we must frame the situation by the logic of the drug’s mechanism. By definition, Paxlovid “works” by stopping replication when the virus needs more of what’s in the unopened bits of Orf1, which are freed by nsp5. This is after three other bits have been freed by nsp3, another protease with a million names. Can you guess what two (potentially all three) of these nsps do? It rhymes with “tintracellular timmune response suppresstion.”
source: zhanggroup.org Nsps 1-3 are released before Paxlovid action pauses progress. Although even more proteins with intracellular-immune-suppression effects will be released later, these two are the “front line” of the virus’s replication process, and may be sufficient to put cellular gene expression (including immune response) on “sleep mode.”
It gets worse. The proteins that are, by definition, still trapped on Orf1 when Paxlovid “works,” are those which normally go on to take up the RNA molecule (before it has degraded, also by definition), and start copying it, and also (by magical RNA rearrangement) make it so that the cell starts to receive and express the portions of the virus’s code that are downstream of the instructions for Orf1, starting with the spike protein. So since Paxlovid stops these proteins from being released, it stops them from taking up the RNA molecule that is being read to produce Orf1s. And so, by definition, it’s possible, if not likely, that Paxlovid leads to more expression of the Orf1 polyprotein.
More expression of the Orf1 polyprotein means more available nsp5 that Paxlovid needs to bind to; and more release of the immune-suppressing nsps 1 - 3.
This is the reason why the virus has ordered events in this manner (if you are a virus, the order of events is defined by which parts of your genes will be read when, which comes down to the physical behavior of the molecules encoding your genes when they interact with other molecules, exactly as for any other life-form). It doesn’t instruct the cell to “make X copies of Orf1,” but to “keep making Orf1 until there are enough that it takes over reading me.” In the meantime, every Orf1 polyprotein is, as a first order of business, releasing a trio of proteins that sabotage the cell’s alarm system. And it hasn’t even begun to let the cell make the proteins that comprise the virus particle itself, so there is little hope of an intervention by the adaptive immune system, particularly if that adaptive immunity is only trained by the spike protein. Pretty smart.
And so this is where Paxlovid pauses things. Exactly when there is as little chance of the immune system taking out the cell as possible.
An Interaction with Innate Suppression, or with the Omicron Siblings?
Even if the anecdotes above are not instrumental to the plausibility of the theoretical argument, is there something about post-trial observations of viral regression after Paxlovid treatment that would fit the theory, and therefor account for why these anecdotes could be signs of an emerging trend? As it turns out, there are two things.
Innate immune suppression: If the Covid vaccines are having a detrimental effect on innate immunity, they may be sabotaging the normal mechanism of Paxlovid, by making it easier for “unpaused” infected cells to resume production and release of complete virus.3
Omicron (BA.1 and BA.2) tissue tropism: Since the Omicron siblings prefer to infect the upper airway tract, “unpaused” cells in both Covid-vaccinated and previously-infected hosts lack nearby T-Cells trained to look for signs of infection. Thus, pre-existing memory immunity (including ramped-up humoral antibodies) does not contribute to suppressing viral regression. Thus, the only way to get functional “Omicron immunity” is to let the infection “play out,” and Paxlovid merely delays this required outcome.4
More in “Unfinished Business”:
Part 1 (this post):
Introduction: Early anecdotes
Official Unglossed Totally Reckless Theory Outline
Theory Explanation
Considerations for Covid-vaccine or Omicron interactions
The original version of this post preceded the theory with the following set of caveats, which are unnecessary in retrospect:
Are these rare outcomes that just happen to have sifted to the top of Chudov’s search query? Artifacts of false-positive antigen tests overlaid with allergy season symptoms? Is Paxlovid actually instrumental in the “trend,” or are the non-treated simply less likely to post their concerns online? The reader may make of these anecdotes what they wish.
What struck me, is how easily I was able to think up a plausible mechanism for how Paxlovid could drive a “(false) recovery, remission” outcome among some portion of recipients. This mechanism struck me as so plausible that it should be considered possible regardless of the current state of the (anecdotal) evidence.
In other words, I realized I should have already proposed this as a danger before any anecdotal evidence came along to suggest it is really happening.
So, the following is meant to be considered as entirely theoretical. This theoretical argument correlates to the anecdotes gathered by Chudov, but neither affirms nor is affirmed by them. It is merely a theoretical argument.
The original version of this post assumed that nsp5 is freed by endogenous proteases. There does not seem to be any research that supports this; and predicted endogenous targets of the nsp5 cleavage motifs do not suggest there would be one (in other words, if our cells produced a protein that likes to chop proteins in the same places as nsp5, it doesn’t seem like the results would be productive). Instead, experiments have demonstrated that nsp5 can self-cleave (as in two copies can cleave eachother) even with an attached head and tail of ~20 extra residues in the N and C domains, albeit at a much slower rate.
For more comments on the significance of the change in tissue tropism in the Omicron siblings, and why this might render therapeutics unproductive, see “RashoCRON.”
I just sat down with a cup of turmeric tea, just to see if there is anything new on Substack, and...
... drumroll...
OMG: THE MOST AMAZING POST showed up! Incredible.
I linked your post to mine and the interplay between them is most interesting and worthy of anyone's close attention.
Just FYI: I did NOT, in any way, selectively choose those reddit paxlovid posts. There is not that many on that subreddit. I always read all paxlovid posts on /r/COVID19Positive because I thought I would find something worth reporting, given Pfizer's reputation, and two things jumped to my attention
1) Horrible taste in mouth, which did not seem newsworthy enough
and
2) The story of Paxlovid NOT helping end the infection, which seemed extremely newsworthy to me, even if it seemed anecdotal and premature.
I am glad this got your interest and there is a cellular mechanism for this sort of turn of events.
I hope that someone very important, besides the two of us of course :-) notices this and tests the Paxlovid recipients more closely.
Just read this and your subsequent articles on Paxlovid. I can't thank you enough for these. I'm a physicist, not a biologist. I actually loathe molecular biology and find something chilling about it. But God bless you for going there. And then putting together your lockpicker sequences for those of us who can't bear to break life down into molecular steps!
A friend had been told by her doctor that this drug would protect her from hospitalization and death. I've offered her ivermectin; her doctor has told her it's elephant tranquilizer or something. She said she'd heard of the fails of Paxlovid but still ...says it's still better than hospitalization and death. Not sure if she'd appreciate your commentaries here but I sure do. Thank you, and thanks to Igor too.



















I just sat down with a cup of turmeric tea, just to see if there is anything new on Substack, and...
... drumroll...
OMG: THE MOST AMAZING POST showed up! Incredible.
I linked your post to mine and the interplay between them is most interesting and worthy of anyone's close attention.
Just FYI: I did NOT, in any way, selectively choose those reddit paxlovid posts. There is not that many on that subreddit. I always read all paxlovid posts on /r/COVID19Positive because I thought I would find something worth reporting, given Pfizer's reputation, and two things jumped to my attention
1) Horrible taste in mouth, which did not seem newsworthy enough
and
2) The story of Paxlovid NOT helping end the infection, which seemed extremely newsworthy to me, even if it seemed anecdotal and premature.
I am glad this got your interest and there is a cellular mechanism for this sort of turn of events.
I hope that someone very important, besides the two of us of course :-) notices this and tests the Paxlovid recipients more closely.
Just read this and your subsequent articles on Paxlovid. I can't thank you enough for these. I'm a physicist, not a biologist. I actually loathe molecular biology and find something chilling about it. But God bless you for going there. And then putting together your lockpicker sequences for those of us who can't bear to break life down into molecular steps!
A friend had been told by her doctor that this drug would protect her from hospitalization and death. I've offered her ivermectin; her doctor has told her it's elephant tranquilizer or something. She said she'd heard of the fails of Paxlovid but still ...says it's still better than hospitalization and death. Not sure if she'd appreciate your commentaries here but I sure do. Thank you, and thanks to Igor too.