The case for inflammation being (mostly) a bystander to Covid-19 pneumonia, not the cause.
Part 3 (this post):
Severe “Covid-19” in turn causes adaptive immune suppression. (go)
Other examples of T Cell suppression and immune “stalemate” in persistent disease. (go)
The likely implications of IgG4 tolerance in light of the reality of severe “Covid-19”: persistent infections, ADE? (go)
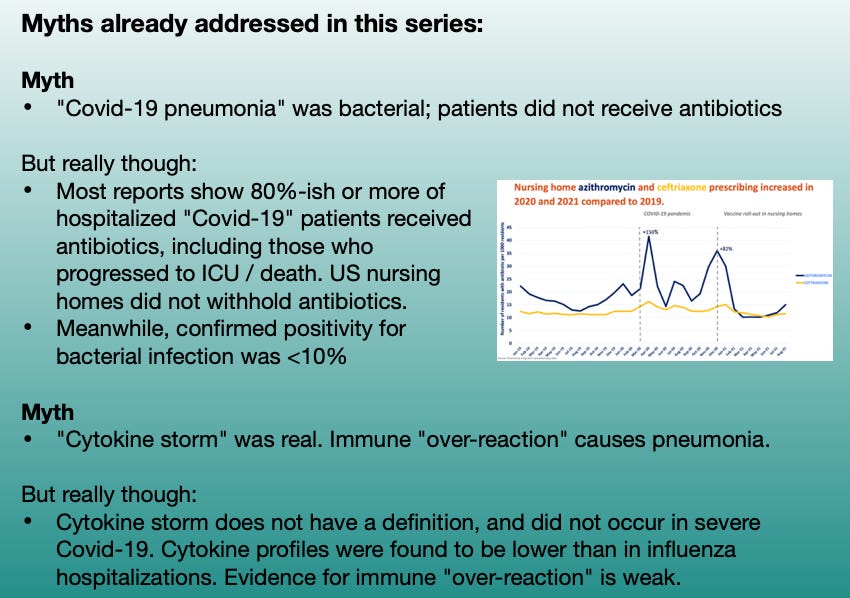
This series, so far, has been 2.5 forays into myth-shattering. It is useful to summarize those myths one more time; as anyone who reads Part 3 without clocking the rest of the series is immediately going to have one of the myths trigger a brain-allergy attack, prompting unproductive “but myth X says!” comments:
When last we were following Shiv Pillai’s talk (embedded below) on the nature of “Covid-19 pneumonia” (Part 1), he was describing how the virus turns off many people’s innate antiviral response using a whole tool-bag of molecular weapons. By shutting off the virus-alarm genes built into every cell (as many, if not all common viruses do), it acts like a metaphorical “invisible pest,” rummaging through the lungs undetected.
With the immune system of older adults asleep at the wheel, the rummaging of the invisible raccoons quickly gets out of hand. Soon the kitchen becomes so trashed with crumbs and turds — things the invisible raccoons leave behind — that the immune system “freaks out.” Cell destruction and viral debris are now everywhere, triggering DAMPS and PAMPS (damage and pathogen-associated molecular pattern receptors), and prompting innate inflammatory responses.
At least the invisible raccoons were polite enough to stand all the waffles on end.
However, as I pointed out in Part 2, there is no strong evidence that this “freaking out” causes the kitchen of our lungs to get trashed in the first place. The virus is doing the real damage. Just as inflammation reacts very strongly to any severe tissue injury, so are the lungs the scene of much post-injury inflammatory response. Certainly there are benefits to modulating this response, including to calm coagulation cascades that synergize with inflammation and can amplify injury; but again this is true of any type of severe tissue injury. The problem is that the immune system allowed the injury to happen in the first place.
Lessons from nature: Adults are not virus-proof
The hazard of viral innate immune evasion is why we build and depend on adaptive immunity formed in childhood; because innate immunity loses function over time.
Pillai discusses at 20:30 how some adults even develop antibodies against antiviral Type I Interferon, predisposing them to severe Covid-19 (these antibodies may in part result from prior generic vaccinations, as a wild speculation on my part; though preexisting cancer or chronic disease may be more plausible). These adults’ immune cells can’t “hear” the antivirus alarm messages even if they go off.
Impairment of adaptive immunity
What is especially notable about this stage of severe disease is that the immune system actively suppresses adaptive response against the virus. Now we may return to Pillai’s talk, starting at 23:45 (queued below):
Ok, I skipped watching that part of the video, what is the next part of the theory?
Please do not skip watching that part of the video. It is integral to the model of disease as two-part immune suppression.
Now, for an overview of what Pillai discusses:
A defect in innate immunity sets you up to get severe Covid-19. […] Once you have severe Covid-19, you also alter adaptive immunity further — so you have an adaptive response, but you’ve altered it […] for a period of time. It is in this window when we aren’t evolving our immune responses appropriately, we aren’t clearing the virus, where we are probably more susceptible for the virus, in the face of suboptimal immunity, to go through antigenic evolution.
How does this suppression play out? As Pillai discusses, the same injury response cytokines that are elevated in severe disease (as they would be after any injury, I would note), suppress the normal, robust adaptive immune response:
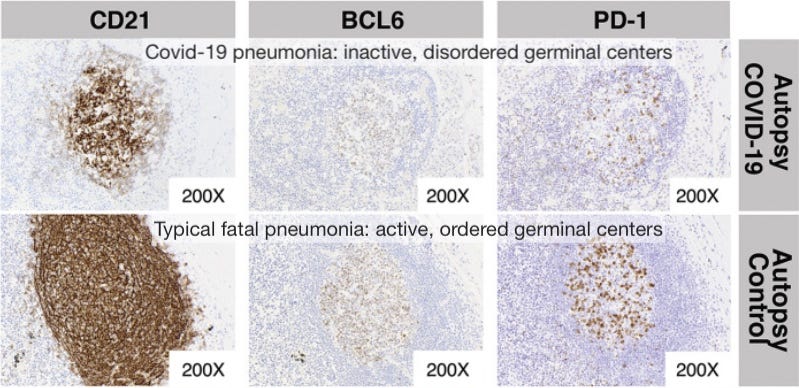
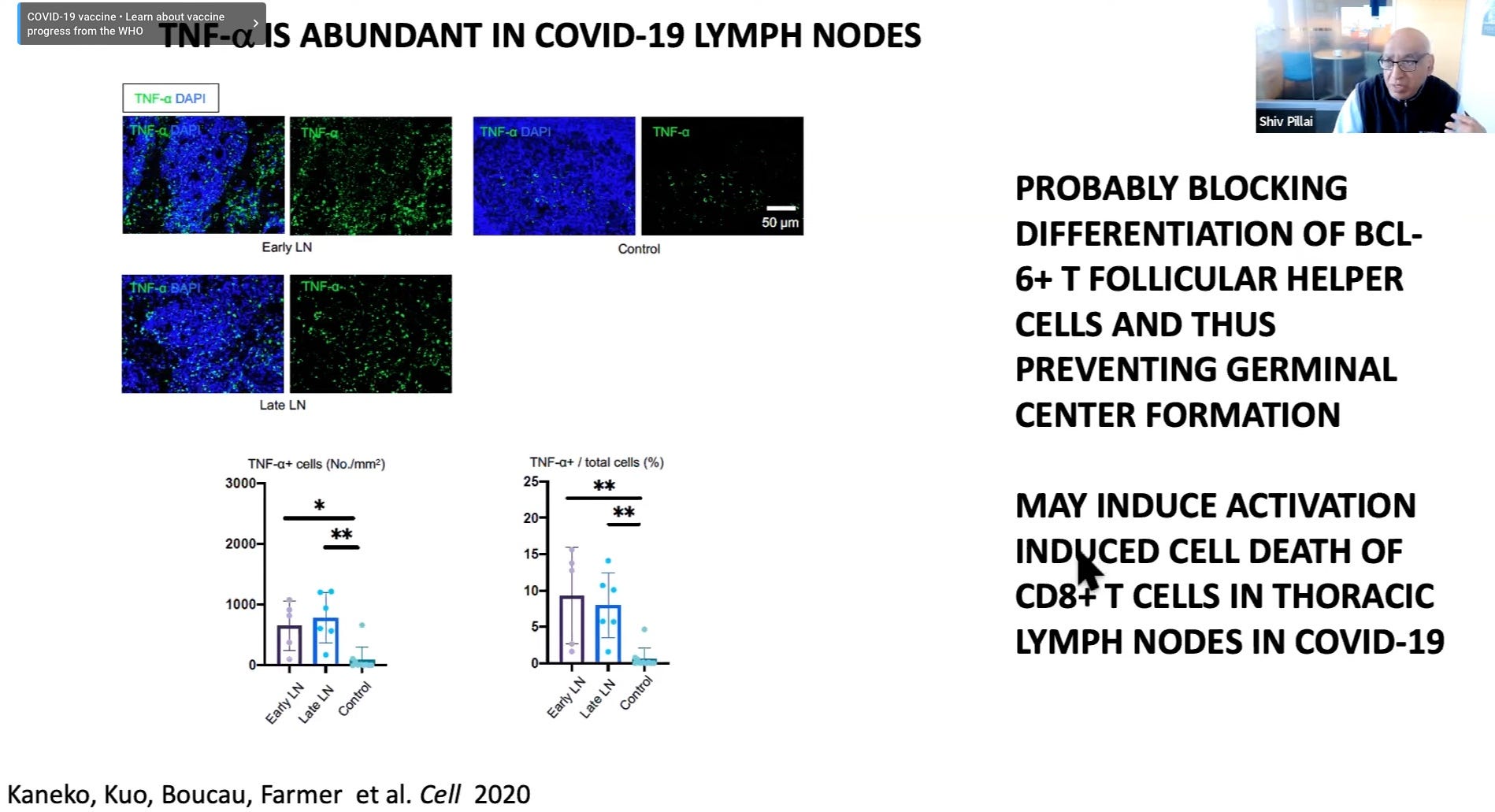
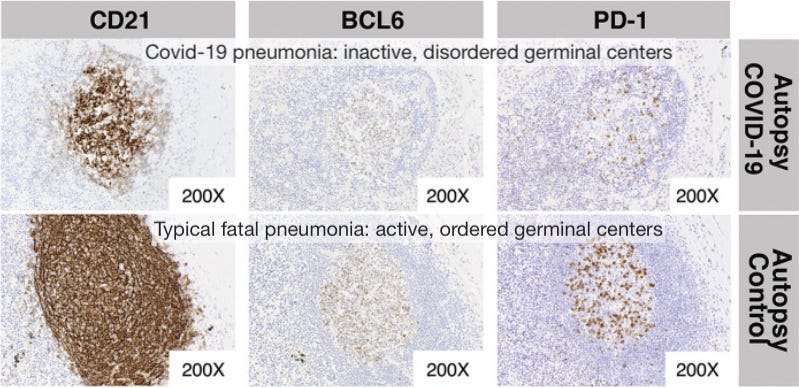
Germinal center freeze-out: Long window in which antibodies do not undergo affinity-maturation (so they aren’t getting better at neutralizing virus). Germinal Center activation (immune suppression) is blocked by high levels of TNF-α.
Here I would note the results observed by Röltgen, et al., in which lymph nodes in autopsies for deaths during severe infections had essentially “clocked out,” showing no sign of active germinal center activity.1
Germinal centers are the “dojos” where B Cells mutate their receptor/antibody-design genes to achieve better binding to the part of the virus chosen by Antigen Presenting Cells for an immune response. Without germinal centers, antibodies cannot get better at binding to the virus; and this effect is observed in severe disease.
CD8+ T Cell expansion is disfavored by the injury response cytokine milieu. Instead of gaining T Cells eager to “seek and destroy” SARS-CoV-2-infected cells wherever they are found, patients in severe disease simply “wave the white flag.”
Bottom: Rather than a “cytokine storm,” a cytokine milieu (TNF-α & β) that suppresses a robust immune response (germinal centers, CD8+ T Cell expansion)
Waving the T Cell White Flag
Further manifestations of innate immune “defect” are detailed in Pillai’s talk, but these two alone are sufficient for observing how rather than being a disease of over-response, Covid-19 is characterized by extreme immune suppression. Adults who fail to mount a prompt innate antiviral response then go on to fail to mount an effective adaptive anti-SARS-CoV-2 response, landing in the 4th quadrant below:
Why would the injury response cytokines add immune suppression “insult to injury”? — well, perhaps to avoid compounding injury.2 The virus has already done enough damage; it makes sense that adaptive immunity would be prevented from unleashing its full fury. (In the kitchen metaphor, the raccoons have now made the kitchen too fragile to use the raccoon-scent-trained “dogs” of CD8+ T Cells to hunt them out; the whole place would fall apart).
The end result is rapid-onset tolerance (even if only temporary) and resulting persistent infection.
In fact, similar dynamics have been observed in many infections; and tolerance in general (i.e. “waving the white flag”) is probably a necessary safety-valve to allow co-existence with many common viruses. John Wherry provides an excellent plain-English description of this dynamic in his recent interview with Amy Proal:
[Besides outcome 1, strong T Cell response eliminating acute infections, and outcome 2, death,] there’s a third outcome [in our generic battle with viral infections], and that’s sort of a stalemate. And, that’s a situation where the virus actually is now mounting counter-attacks against this immune response.
At some point, at around the two-week mark in both animal models and in humans, it seems that the immune system has an evolutionary choice to make. If it keeps mounting this really aggressive response, killing infected cells, you’re going to end up damaging infected tissues so much that the host dies, not just because of the virus but because of the response to the virus.
[…] The immune system decides to stand down, at least partially — go into a state of deciding that this is going to be a long-term battle, we need to figure out how to contain this invader; we can’t fully eliminate it. And T Cell exhaustion is one strategy that allows that to happen. The T Cells reduce their activity, they stop their all-out war on the tissues, but they maintain some ability to constantly monitor, basically form a stalemate, constantly pushing especially herpes-type viruses or other sorts of persistent viruses into a state of containment.
And indeed, multiple studies have found T Cell responses in severe disease to be characterized by suppression and exhaustion — the T Cells, like germinal centers, have “clocked out.”3
IgG4 Tolerance: Persistent Infection Guaranteed, and eventual Antibody Dependent Enhancement?
At last, we arrive at the topic allegedly promised by the title of this series. What can be expected from future infections as the mRNA-transfected, especially those still boosting after dose 2 (which already begins IgG4 conversion), become more and more primed for the virus?
We can make some wild guesses of what is “likely,” even though very little has been confirmed about post-“breakthrough”-infection IgG4 dynamics:
Higher baseline likelihood of persistent infection.
Feedback-loop increases in persistent infection susceptibility (each infection ramps up IgG4), resulting in a “persistent infection sink” effect.
T Cell exhaustion will also ramp up over time, as suggested by the recent study “super-boosting” mice with a non-mRNA vaccine.
Lots of viruses seem to form persistent infections, including from childhood; and as discussed in Wherry’s talk, tolerance may be a good thing at that point, not bad.4
However, the following dynamic really does not seem like a good “trade”:
Kids, including infants, who were never at apparent risk for (severe disease leading to) persistent infection, now must run a decades-long gauntlet with a potential, yet-to-be-quantified propensity toward persistent infection. If propensity (i.e. IgG4 and T Cell exhaustion) only increases with each exposure to the virus, the true implications won’t be understood for decades (assuming any of these kids’ hearts can go that long).
In general, I remain of the opinion that tolerance-enabled persistence for this particular virus is bad, not good; and will increase susceptibility to Long Covid and post-viral cardiac harms:
As a final comment regarding Pillai’s theories on persistence and variant-generation: This is another open question. Whereas Pillai alleges (echoing vanden Bossche, of all people) that the "suboptimal," germinal-center-defective antibody response is likely to create an escape pressure environment, it’s just as plausible that high-affinity IgG4 antibodies will accelerate evolution even more. However, it should be mentioned that we aren’t even sure whether coronaviruses actually evolve to escape immunity that much.5
But what about tolerance and severe disease?! It is literally what is in the title, Brian
Ah, yes.
The entire point of critiquing the common understanding of severe Covid-19 as a “storm” of immune “over-reaction,” is to suggest that IgG4-mediated Antibody Dependent Enhancement may begin to occur.6
If the Covid vaccines almost certainly protect against severe outcomes in part by tagging infected cells for destruction, IgG4 will sabotage that benefit. This may replicate the original problem that led to severe Covid-19 to begin with; namely, suppression of innate response to cell and tissue infection:
Tolerance in light of a correct understanding of severe Covid-19:
Suppressing immune response to infected cells causes and is caused by Covid-19 pneumonia.
Tolerance suppresses immune response to infected cells.
Oops.
This could be a problem even if IgG4 still “limits” viral dissemination to other cells thanks to high binding affinity (unlike the antibodies made in natural severe infections, IgG4-making B Cells have spent ages mastering their binding affinity in germinal centers).
As far as I see it, it is all a question of degree, of balance. Right now, the balance still seems to be in favor of preventing the virus from turning the lungs into raccoon-trashed kitchens. But, especially given that IgG4 can interfere with IgG1 in heterologous complexes, that could all change as soon as a certain threshold of IgG4 conversion is met.
If that happens, the Covid-19 vaccines will have wound up deputizing the raccoons.
Additionally, high concentrations of DAMPs and injury-associated cytokines may suppress germinal center and T Cell expansion to guard against acquisition of autoimmunity in the wake of injury (as injuries will flood the corresponding tissue with self-antigens).
This outcome would not match any previous traditional applications of the term. However, it seems like an apt characterization of severe disease resulting from IgG4 immune suppression.
I'm starting to really appreciate Brian Mowrey's contribution to this whole Covid-19 topic. His explanation as to the myth of the cytokine storm (as opposed to DAD) was welcomed. Now, I wish I have more of a serviceable grip on "all-things virology and vaccinology" but...
Great series of posts Brian! It's probably tough catching flak from both sides, but ultimately I think this process is producing the strongest arguments.
Regarding testing, especially in the US, almost all testing is nasal swabs which is of the mucus membranes. My understanding is IgM dominates the response there. It seems that response will be identical in both Vaccinated and Unvaccinated. If the immune tolerancing theory is correct and the Vaccinated start having persistent infections, would it continue to show up in the nasal swab tests or would anal swab tests or blood serum tests be more accurate?
Anecdotally, I have seen a case where a "vaccinated"19 year old healthy BMI female has had a persistent cough for 1.5 months. Tests "negative" for Covid with a nasal swab and has no other symptoms. I also see plenty of anecdotes about persistent viral infections "but it's not covid". Is it possible that we are already seeing persistent infections but our testing regime is failing us? Keeping in mind of course that there really are other viruses out there that we routinely do not test for.















I'm starting to really appreciate Brian Mowrey's contribution to this whole Covid-19 topic. His explanation as to the myth of the cytokine storm (as opposed to DAD) was welcomed. Now, I wish I have more of a serviceable grip on "all-things virology and vaccinology" but...
Great series of posts Brian! It's probably tough catching flak from both sides, but ultimately I think this process is producing the strongest arguments.
Regarding testing, especially in the US, almost all testing is nasal swabs which is of the mucus membranes. My understanding is IgM dominates the response there. It seems that response will be identical in both Vaccinated and Unvaccinated. If the immune tolerancing theory is correct and the Vaccinated start having persistent infections, would it continue to show up in the nasal swab tests or would anal swab tests or blood serum tests be more accurate?
Anecdotally, I have seen a case where a "vaccinated"19 year old healthy BMI female has had a persistent cough for 1.5 months. Tests "negative" for Covid with a nasal swab and has no other symptoms. I also see plenty of anecdotes about persistent viral infections "but it's not covid". Is it possible that we are already seeing persistent infections but our testing regime is failing us? Keeping in mind of course that there really are other viruses out there that we routinely do not test for.