


Repeating the case against bacterial coinfection (CSA)
For readers who missed the first round

This post will reply briefly to a fresh repackaging of the zombie theory that deaths attributed to “Covid-19” in 2020 and afterward were some mixture of misclassified common deaths and and untreated bacterial pneumonia, this time by Neil, Hockett, Engler, and Fenton. By necessity it will repeat points I have previously raised, so it is geared more toward readers who have not already seen the theory challenged here.

This suffices for an introduction; we may now get to the points.
i. Defending the different-ness of Covid-19 (some new content, as well as discussion of the distinct problem of blood going to airless-lung-regions discussed here)
ii. Patients were given plenty of antibiotics, so it is nonsense to suggest they died from bacteria. (previously shown here).
iii. Loose ends.

i. Different, distinct, and deadly
What is different about inpatient “Covid-19”
Neil et al. highlight that one of the signature clinical traits in severe Covid-19 is not unknown in influenza and other forms of pneumonia:
Chest radiographs were abnormal with air-space shadowing such as ground-glass opacities. However, this is not unique to covid-19. Ground glass opacities are also commonly associated with influenza infections and with a wide range of other lung conditions, including pneumonia.
From these early notes from Wuhan, Neil et al. pass to Italy, where an emergency department head informed the news expressly that Covid-19 is not like flu.
“No, it’s totally another thing. I think it is (mumbles) pneumonia because most people get pneumonia, and as I have said before it is a very severe pneumonia…
The reader it would seem is asked to understand that the speaker is so over-eager to distinguish Covid-19 from flu that he misarticulates, i.e. that when he says “it’s a very severe pneumonia” he means to say that it is common pneumonia, which is more severe than flu. Therefore Covid-19 is not like flu because it isn’t special at all, in fact it is just regular pneumonia exacerbated by panic. His alarm and distress all stem from the dire consequences of over-reacting to a normal, daily task category. This is a not a particularly supportable interpretation.
To a certain extent the argument in Neil et al. is linguistic and deconstructionist; just as one can show the difficulty of defining “woman” in terms that individual people called “woman” might not meet, Neil et al. challenges the notion that “Covid-19” has a meaning distinct from common pneumonia. At the same time, it regularly insinuates that in fact there is a distinct meaning, but that it derives exclusively from the human reaction to the application of the term, subsequent a positive PCR test. All these excess deaths, the full ICUs, the dead grandmas — they stem not from having X but from detecting and saying “X.”
For purposes of grounding things back into the practical and material, a simple way to think of this is that so long as being positive for SARS-CoV-2 distinguishes pneumonia patients from “pot luck” pneumonia patients in any sense as a group, then there is a quality to pneumonia associated with SARS-CoV-2. Certainly this quality could be iatrogenic, i.e. related to medical over-reaction, but it also could be related to the virus. There isn’t such a particular need to harp on the linguistic argument. In fact this really just serves to dislocate the reader, to distract them from the pure and implacable material fact that pneumonia associated with SARS-CoV-2 isn’t like pot-luck pneumonia.
It has qualities. Again, leaving aside the question of treatment vs. the virus, it was more severe. Patients going to the ICU for pneumonia “with Covid-19” stayed much longer than those going to the ICU for other causes. I will pin most of this post to Gao, et al., the recent “cytokine storm is not real (true), most patients died from secondary infection (false)” paper. 190 patients experiencing respiratory failure “with Covid-19” were compared with 395 patients experiencing respiratory failure for any other cause.
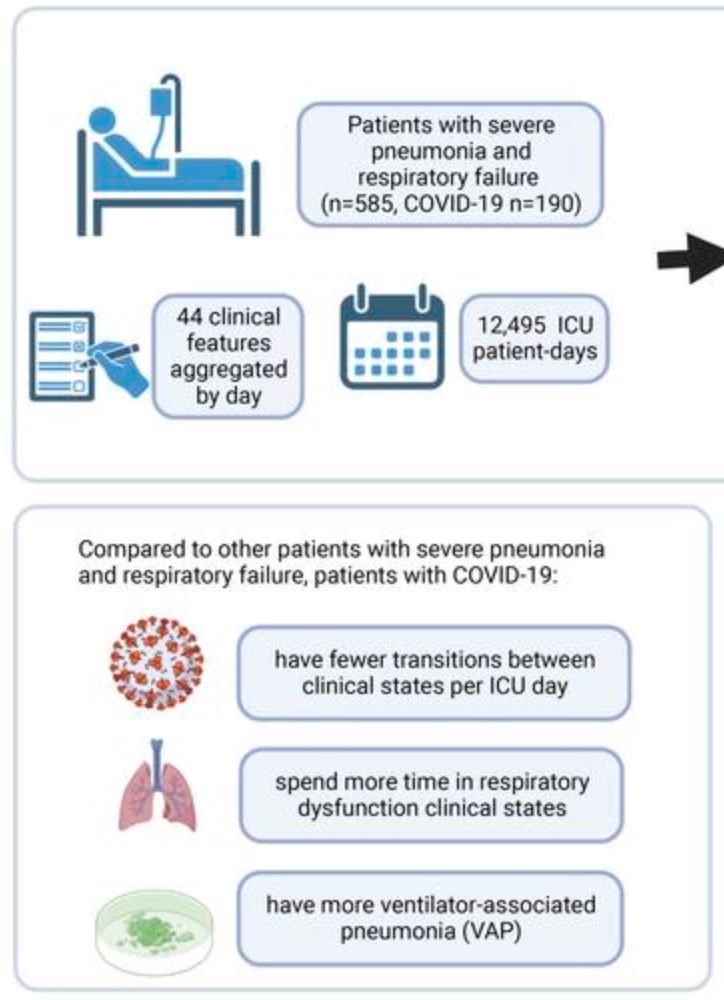
The patients with Covid-19 spent longer in the ICU. They spent longer in the most critical and high-mortality conditions. Therefore, being positive for SARS-CoV-2 predicts and detects worse outcomes than regular respiratory failure. Either because the virus leads to a more severe pneumonia, or because the positive test leads to maltreatment. Below, the width of rectangles corresponds to the average days spent in any given clinical state, and the states on the right are colored more red in accordance with higher mortality.
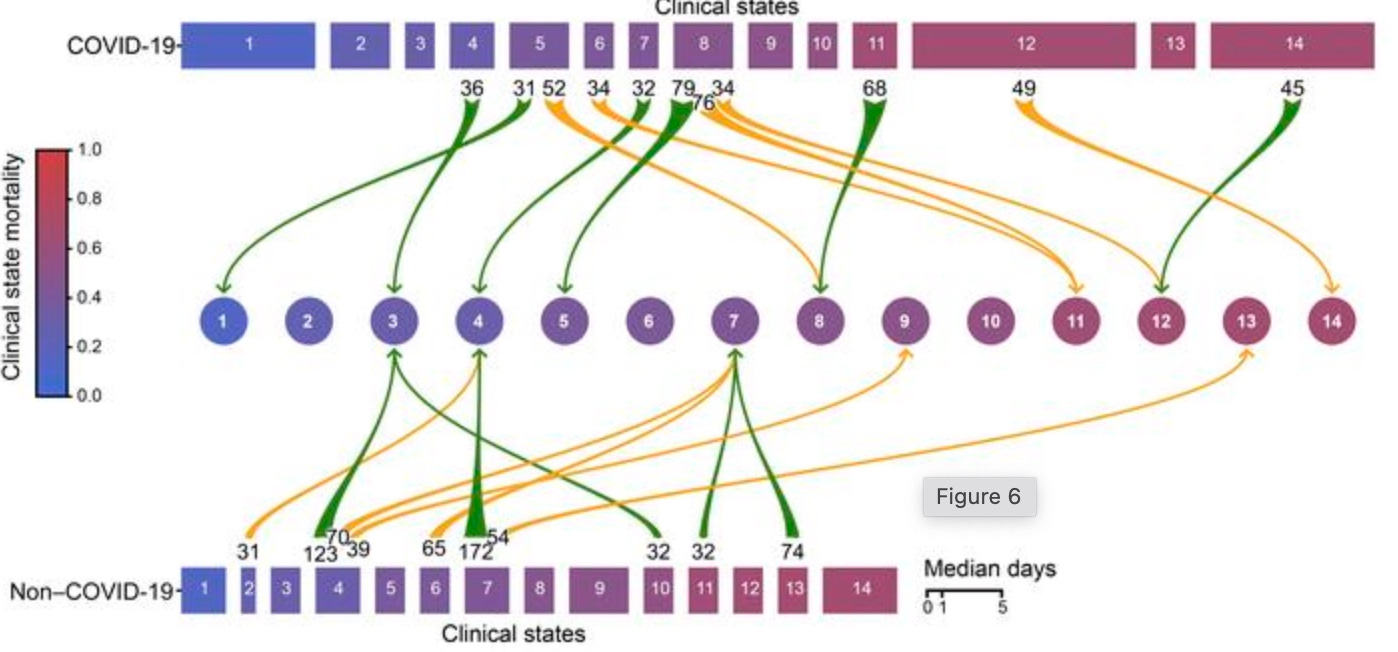
In this same way we can understand the clinical features associated with SARS-CoV-2 materially, steering clear of linguistic sand-traps. Ground-glass opacity is more common for patients who are being cared-for “with Covid-19” (respiratory distress presumed to be related to SARS-CoV-2 via a PCR test) than patients defined by any other biological factor one can come up with. Pathologically, patients who die “with Covid-19” have manifestations of diffuse alveolar damage and organizing pneumonia more often than people who die with any pot-luck group of conditions.1 Defining a group of patients or deceased in terms of being positive with the virus predicts differences from regular pneumonia. If one merely wants to argue that iatrogenic harm causes these differences, there is no need to shove them under a rug at the same time. This confesses a need not to really let the reader see the differences.
What is distinct about inpatient “Covid-19”

All of this is to grant the argument in Neil, et al. the most generous reading possible. Let’s say that all that distinguishes “Covid-19” is a positive test and a tendency for group outcomes to be different. Then the authors would still need to wage a positive case that these differences result from aggressive ventilation of people with regular pneumonia. And it would be fair to grant that such a positive case is difficult to litigate, since only a handful of the hundreds of thousands of inpatient experiences have been published in case report format.
This is the framing assumed by Neil, et al., but it isn’t what actually prevails. Respiratory distress associated with SARS-CoV-2, i.e. “Covid-19,” was distinct from any other form of pneumonia from the very start in a clinical forms which received less attention in the mainstream media than “ground glass opacity.” It was unlike common pneumonia not only in group characteristics but individually, with a high prevalence of both clotting disorders and even more-so of hypoxia related to pulmonary perfusion of airless regions of the lung. Pneumonia per-se wasn’t really what drove respiratory distress in Covid-19, but rather the fact that patients’ lungs were allowing blood to flow to regions that were flooded with fluid and absent of air. This is an important technical point of which the Bacteria Theorists seem totally unfamiliar, but which the reader can master for themselves with a little effort.
The arteries of the lung contract by reflex when the tissue they serve does not have oxygen. If 30% of the lung is unventilated, and the arteries serving this region contract as they should via the HPV reflex, then the blood leaving the lung will still be 100% oxygenated. Patients with Covid-19 were observed to experience a failure of this crucial reflex — blood still went to airless regions, and as such in the example above the blood leaving the lung would be only 70% oxygenated. This is the distinctive hypoxia of Covid-19; it was recognized from the start in Italy, but received little attention as a biological phenomenon until Archer, et al. showed it to be related to the proteins programmed by SARS-CoV-2 and coronaviruses.
From Italy, in early 2020:2
COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome
A possible explanation for such severe hypoxemia occurring in compliant lungs is a loss of lung perfusion regulation and hypoxic vasoconstriction.
And regarding the recent study explaining these results in terms of viral etiology:

Covid-19, in short, is not like regular pneumonia. While ventilation may be detrimental or in some sense a necessary evil in treatment, it is insufficient to explain this distinct failure of the HPV reflex and resulting extreme hypoxia.
What is deadly about “Covid-19”
The final problem with the Bacteria Theory is the following. Again, this is to show why the generous interpretation that “Covid-19” is only different from other pneumonias in terms of group differences, and maybe these group differences stem from mistreatment, is unsupportable: If Covid-19 was only more severe because of mistreatment (excessive ventilation), what explains why patients turned away with a positive test (as per the protocols in place for most of the time in question) came back to the hospital days later in respiratory distress? The raw hospitalization rate of Covid-19 (i.e., the rate of hospitalization following a positive test) exceeds what would follow any other biological agent even without antibiotics and cannot be explained by whatever happened after hospitalization. It totally defies any explanation from the Bacteria Theory. It is like some confusing hallucination; except in fact it is reality, and demands that the model of reality proposed by the Bacteria Theory be questioned.
And, again, why so many?
What Neil et al. propose is that SARS-CoV-2 is only one of “approximately 200 respiratory viruses that can lead to respiratory disease,” but that in the case of that one, as in when it was detected on a PCR, patients entering the hospital for what should have been routine pneumonia treatment were over-ventilated. Even if the iatrogenic response were 100% lethal, we would expect a mere addition of .5% of annual pneumonia hospitalizations to the annual pneumonia death count.
For US adults, pneumonia is the most common cause of hospital admissions other than women giving birth.
About 1 million adults in the US seek care in a hospital due to pneumonia every year, and 50,000 die from this disease.
So, per Neil et al., we propose that 1 out of 200 of these 1 million adults hospitalized for pneumonia have the innocuous biological agent that condemns them to (for purposes of example) a 100% lethal medical response: We thus produce 5,000 extra deaths 3 years in a row. Needless to say, this is vastly short of the excess deaths experienced by the US in the years in question (essentially equivalent to figures for “Covid-19 deaths” — 420,000 per year). It isn’t even in the ballpark.
Bearing in mind again that patients were by policy only hospitalized after they were already in respiratory distress, and bearing in mind that patients hospitalized for “Covid-19” actually mostly lived through it (e.g., 82% of hospitalized patients in the Paxlovid trial placebo arm survived), what would be necessary for a PCR+ of SARS-CoV-2 to be so deadly if only because of mistreatment? It would need to be true that SARS-CoV-2 caused an average many times over 100% of all “regular” pneumonia hospitalizations throughout 2020 and 2022 (and we can model this as at least remotely possible due to failure to use antibiotics before hospitalization). Simultaneously, it caused an extraordinary amount of “regular” pneumonia hospitalizations in the summer of 2021, even though this isn’t regular at all. Simultaneously, per the Bacteria Theory, it is normal for SARS-CoV-2 to cause this many hospitalizations; it was doing so all along; in fact it has always been the case that over 100% of people hospitalized for pneumonia are positive for SARS-CoV-2, we just never bothered to make a big deal about it before (but, if really so, why not?).
So, essentially, it would mean that all pneumonia is accompanied by SARS-CoV-2 PCR-positivity and always has been, but this fact is simultaneously of no relevance to anything or anyone.
ii. Hospitalized patients received antibiotics
There is little to add here to my previous report on the subject:

If patients dying with Covid-19 pneumonia were in fact dying of bacterial pneumonia, then some improvement should have been expected from the fact that actually they were almost all given antibiotics, regardless of over-ventilation. Even more problematic of course is the fact that actually they didn’t test positive for bacterial infection.
Among 213,338 Inpatients with “Covid-19”*
All, received antibiotics: 77.3%
Ventilated, received antibiotics: 95.9%
Died in hospital, received antibiotics: 91.7%
*primary or secondary ICD-10-CM B97.29 February–April 2020 or primary or secondary U07.1 (COVID-19) April–October 2020, 716 hospitals3
Antibiotics were used “empirically,” i.e. in absence of PCR or culture-confirmed bacterial infection; but they didn’t improve outcomes in any apparent manner. Altogether, secondary infections were uncommon (either because of or regardless of abundant antibiotic use):
For COVID-19, 62/806 (8%) patients were reported as experiencing bacterial/fungal coinfection during hospital admission. […]
Despite frequent prescription of broad-spectrum empirical antimicrobials in patients with coronavirus-associated respiratory infections, there is a paucity of data to support the association with respiratory bacterial/fungal coinfection.4
This is an important point as far as a simplistic or straight-forward approach to the question of whether Covid-19 deaths were caused by bacterial pneumonia which was denied treatment. “Ok, did someone at some point test whether Covid-19 patients had bacterial infections, or report whether they were given antibiotics?” → “Yes, these things were tested and reported.” → “No, they didn’t have bacterial infections, and besides were abundantly given antibiotics.” → “Ok, this theory is wrong.”
And so here we explain the resort to complicated models in Neil, et al., which I have not addressed in full scope because none are facially sufficient to answer why 420,000 American excess deaths per year.
I’ll add one small note regarding overall antibiotic use. If it is known that Covid-19 patients were overwhelmingly prescribed antibiotics, it is not the case that testing positive for SARS-CoV-2 reduces the likelihood of receiving antibiotics, and therefore it cannot explain the overall reduction. So the overall reduction must simply be explained as resulting from the broader social response. In particular, it should be remembered that a great deal of antibiotic prescriptions are given gratuitously in advance of surgery and have no relationship to infections or illness to begin with; and surgeries were drastically reduced in response to the virus.
iii. Loose ends
With the above objections raised, there is no need to be comprehensive regarding the points raised in Neil, et al. I’ll only note two more issues.
The pneumonia vaccine study
Neil, et al. write:
Lewnard et al looked at the interaction between bacterial pneumonia and SARS-CoV-2 and found some interesting results, especially when investigating whether vaccination for pneumonia reduced the risk of covid-19 disease. [they find it does]
This is a problematic study in that it takes place in early 2020 in California, when there was no widespread outbreak as in the east coast and elsewhere. Therefore it is measuring a rare outcome, which makes it more likely that noise will overcome signal. There were only 1,075 hospitalizations and 334 deaths among 531,033 enrollees; additionally, only 80,600 of the same enrollees had failed to receive the pneumonia vaccine in question. For this reason a literal handful of deaths in the “control” group, if explained by some bias related to not receiving a pneumonia vaccine, would drive the result; and this bias might not be related to the overall bias for the groups as a whole (it only corresponds to a subset of individuals not receiving the vaccine). This subset was probably those individuals in the insurance system already in terminal care, who would no longer be keeping up on their vaccines. This explains why the same “protective effect” was found for receiving herpes zoster vaccine in the same study. Notably, individuals in the pneumonia vaccine group received antibiotics just as often (as a whole) as those without the vaccine; so it is unlikely that the vaccine did much to protect against bacterial infections to begin with. Pneumonia vaccines, like most, probably don’t keep anybody alive.5
The epidemiology
So much of Neil, et al. is devoted to epidemiological arguments that are imposing and bewildering. I will only say a little, repeating points I have made previously.
Adult and elderly humans depend on memory immunity, whereas children rely on extremely robust innate immunity while they build the memory immunity that will protect them in the future. To argue that a novel virus, to which no adults possess immunity, in theory can’t cause excess deaths, especially among adults, is to assert that memory immunity serves no protective function in adults. Well then, what is it for?
Coronavirus genomes code for multiple sophisticated immune suppression genes which we barely understand. Severe Covid-19 is a disease of immune suppression, demonstrating exactly what we would expect if we took what we know about coronaviruses and removed pre-existing adult memory immunity (antibodies and T-cells). To propose a mystery here is to confess either ignorance or disagreement with virology and immunology. This is fine, but the tone of Neil, et al. frequently suggests that what is observed disagrees with what was expectable per existing knowledge. It does not.
This misrepresentation of the state of knowledge is often reflected (either in this paper or elsewhere) in comments regarding the geographic and temporal irregularity of the virus’s spread in 2020; or the fact that the virus did not disappear or attenuate after the first year.
But neither of those things were actually unusual. They were observed and remarked upon in all three of the 20th Century flu pandemics.
The behaviour of influenza has seemed so erratic as regards its occurrence in both time and space that it has fascinated epidemiologists for many years
—Andrewes, 19536
A fact little-remembered today is that novel influenza outbreaks were often so geographically capricious that it was unsupportable for anyone observing propagation of the illness to claim that person-to-person spread is the exclusive means of the same. Reviewing the 1957 flu and three prior pandemics, Shope writes:7
However, certain discrepancies enter to spoil the perfection of the case-to-case transfer explanation for the spread of influenza during the second wave of the 1918 pandemic. These have to do with certain flukes in distribution, certain skips of large bodies of population. For example, Boston and Bombay had their epidemic peaks in the same week, while New York, only a few hours by train from Boston, did not have its peak until 3 weeks later(10). In like manner, Seattle, Los Angeles, and San Francisco had their epidemic peaks some 2 weeks earlier than Pittsburgh, which is just an overnight run from the infected eastern seaboard cities. In some respects, the epidemiologist had an easier time getting the pandemic disease transferred over long distances than in taking it to communities nearby. Thus, though it got to Chicago, presumably from Boston [but Shope really means to say that nothing of this nature can be presumed at all], fairly early and affected that city in September, it did not reach Joilet, just 38 miles away, until October. Similarly, it took 3 weeks to cross the little State of Connecticut from New London County to Fairfield County.
This is just to ground our expectations; any claims about how viral outbreaks “should” spread, i.e. what we should “expect” of them, warrant absolute skepticism.
And so, while the autumn wave of 1918 was one of many examples of rapid, global diffusion of influenza A, the spring and summer-time outbreaks of H2N2 in 1957 offer precedent for a more limited and elusive outbreak. Before I quote from Shope again, let us review what Neil, et al. claim about spring, 2020:
The pattern of spread, and the geographical concentration of the Covid-19 mortality toll is not what one would expect from a respiratory virus. Deaths were highly localised in care homes and hospitals, within elderly age groups and those suffering multiple comorbidities and in specific cities and regions such as New York, Lombardy and Madrid.
For instance, by May 2020 the 'pandemic' in the USA had only occurred around a few points that could have been pinned on a map, and everywhere else failed to experience it.
Buzzer sound-effect, wrong, especially for an off-season outbreak of a novel virus. In summer, 1957, again per Shope:8
In this country, the disease spread slowly, involving initially military establishments that had received personnel returning from the Orient. It appeared in various groups of civilians that congregated from different parts of the United States during the summer, most notably in summer camps and in a summer church conference at Grinnell, Iowa. Individuals returning from these meetings set up foci of infection in their home communities, and by late July and early August the disease was widely seeded throughout the United States. During the early part of the outbreak, Asian influenza showed little tendency to spread except on very close contact and tended to remain sporadic. With the beginning of autumn, the disease diffused more widely and rapidly than it had at first(1).
Thus 1957 is almost a perfect precedent for 2020; except that even in the winter, 2020 and summer, 2021 outbreaks remained contact-limited and sporadic. The transition to true “wide and rapid” diffusion was marked and explained by the extreme genetic modifications of Omicron, which retroactively explain the limited and sporadic nature of the virus’s spread before Omicron. This is another fact that is “hallucinatory” if one is trying to understand reality according to the Bacteria Theory model. What about the mutations to SARS-CoV-2 that gave us Omicron changed bacterial pneumonia?
Meanwhile it is true, as acknowledged elsewhere in this post, that in 1918 secondary bacterial infection was observed to precede deaths. But once antibiotics became available, flu pandemics still happened, and people still died, either observably from post-infection pneumonia or from cardiovascular and other diseases — not quite exactly as was experienced between 2020 and 2023, but rather close. The picture in 1957 was of pure influenza pneumonia in children, a mixture of bacterial and viral pneumonia in adults, and influenza complicating existing diseases.
The research carried out at all stages of development [of influenza in children] showed that the second phase of the disease was connected, not with a secondary bacterial infection, but with the influenza virus.
Under the influence of influenza, mortality from other diseases, the course of which is affected by influenza, rose somewhat. Thus in October and November 1957, compared with the same months in 1956, mortality from diseases of the cardiovascular system rose by 23.8% and from tuberculosis by 46.1%.
—Zhdanov, 19579
Finally in the H3N2 outbreak, it was unexpectedly found that the virus was more infectious and more severe in the second year in the UK.10

Influenza pandemics are the only precedent we have for respiratory viral pandemics, and they have never been regular or predictable geographically or temporally. There is a world of literature warning the modern “detective” not to stare into charts and look for secret patterns. Viruses are wild.
The Bacteria Theorists will wave statistic after statistic after statistic at the reader and demand an explanation for “discrepancies” to whatever their mental model of a pandemic is, but never both to actually read a single report regarding previous pandemics. It’s actually weird at this point.
SARS-CoV-2 is indeed unprecedented in that it is the first recorded novel respiratory virus to “do a flu pandemic” that wasn’t actually Influenza A. I have offered a theoretical justification for this particularity in a previous post; namely, that SARS-CoV-2 may be a recurring ancient virus that simply went unnoticed in prior outbreaks due to humans being older and unhealthier today than ever in the past.

Conclusion: The need to reexplain the explained
The idea that Covid-19 deaths were driven by bacterial secondary infections is seductive because it fulfills the contemporary (American-media-induced) need to discover that nothing is simple; everything has a secret dimension which once discovered allows one to feel clever. “Vacuums do not suck air, they blow it.” “Infection with SARS-CoV-2 does not kill, rather it is secondary bacterial infection.” Even treated bacterial infection is therefore proposed to be the real culprit behind deaths in the mainstream media interpretation of studies that support no such conclusion (i.e. that show critical patients without coinfection dying at almost exactly the same high rate as those with)


If the above study can find that mortality is not different in patients getting bacterial pneumonia while on ventilation, suggesting naturally that bacterial infections make little to no difference in critical outcomes, and yet trigger headlines claiming that secondary infections drive most “Covid-19 deaths”(!), the reader will hopefully not find outlandish the suggestion that people’s brains just really need it to be the case that there is some secret “real” reason why people infected with SARS-CoV-2 got sick and died.
This need extends retroactively and poisons naturalistic interpretations of historical events. Much is made of the statistical habit of tracking pneumonia and influenza deaths as a single metric; of the fact that yes, secondary bacterial pneumonia did in fact precede death in the 1918 flu; and etc. for other nuances, that simply put do not meet the standard of epidemiological rigor upon which historical interpretations of events were based the first time around (I do not speak of “epidemiological rigor” in terms of modern publications, especially of Spanish Flu revisionism which continually raises old death counts). Often books’-worth of careful and nuanced analysis are left un-consulted and in place of a true reassessment of this wealth of evidence, one or two quotes about aspirin are presented and then a mic drop. A complex past carefully exposited is erased with a lazy modern simplification. It is as if an appeals court upturned a decided case because a lawyer showed up with two pages from the defendant’s argument that were already aired in the first trial anyway.
With that said, I acknowledge that in the case of SARS-CoV-2 there has not yet been anything like a careful and thorough epidemiological assessment of the entire phenomenon. The “default narrative” is beset with absolute confusion, and for this reason critical analysis is useful for dispelling misapprehensions in the same. However, when one looks at the question of bacterial coinfections, it can only lead toward a more firm resolution that the virus is what kills.
If you derived value from this post, please drop a few coins in your fact-barista’s tip jar.


Informal citations, since I am working on mobile:
Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7674971/
Organ manifestations of COVID-19: what have we learned so far (not only) from autopsies?https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8975445/
Rose, AN. et al. “Trends in Antibiotic Use in United States Hospitals During the Coronavirus Disease 2019 Pandemic.” Open Forum Infect Dis. 2021 Jun; 8(6): ofab236.
Rawson, TM. et al. “Bacterial and Fungal Coinfection in Individuals With Coronavirus: A Rapid Review To Support COVID-19 Antimicrobial Prescribing.” Clin Infect Dis. 2020 May 2 : ciaa530.
See also:
Hughes, S. et al. “Bacterial and fungal coinfection among hospitalized patients with COVID-19: a retrospective cohort study in a UK secondary-care setting.” Clin Microbiol Infect. 2020 Oct; 26(10): 1395–1399.
Adults already have humoral immunity to bacteria; the whole concept of vaccinating them for this is absurd.
Andrewes, 1953, “Epedimiology of Influenza.”
See also, regarding flu irregularity:
All of chapter 1 in Jordan, Edwin O. (1927.) “Epidemic Influenza: A Survey.” Archived online at https://quod.lib.umich.edu/f/flu/8580flu.0016.858
Andrewes, 1949, “Influenza in Perspective.”
Payne and McDonald, 1957, “Symposium on the Asian Influenza Epidemic.”
And Shope, Zhdanov, and Miller, et al. below
Shope, 1958, “Influenza: history, epidemiology, and speculation.”
ibid.
Miller, D. L. et al. (1971.) “Epidemiology of the Hong Kong/68 Variant of Influenza A2 in Britain.” British Medical Journal. 1971 Feb 27; 1(5747): 475–479.
I'm not trying to be a contrarian here, but your theory runs counter to what I saw in patients, both pre and post Pneumonia development, that were given antibiotics for their positive SARs-CoV-2 test results and symptoms, versus what I saw in COVID patients who were not given antibiotics.
I saw a higher survival rate in patients given the antibiotics, both before & after pneumonia development. Now this is not a huge number I personally witnessed; but the outcomes were so strikingly disparate, that I find the secondary infection hypothesis quite plausible.
This is an active area of discussion and disagreement, but what I find most disconcerting is the lack of constructive engagement from the authors (Engler & Hockett) re. the evidence that Covid-19 ARDS is unique and distinct in several important ways from bacterial pneumonia ARDS. I pointed to this evidence on the unabridged substack version and that evidence was dismissed as having been 'debunked' - a comment liked by Neil and Hockett and approved of by Engler in a reply which didn't appear to make much sense to me. I asked for evidence of the 'debunking', pointing to McCullough supporting the unique clinical pathology of Covid-19 but the response from the commenter was that McCullough was wrong. That was it. No further constructive engagement either from the authors or that dismissive commenter. Not really helpful.