


The SARS-CoV-2 and Blood Going to Air-less Lung Areas Study
How SARS-CoV-2 sows confusion in the vasculature of the lung (follow-up to the Kids' Lungs Study).

A new study illuminates a possible mechanism for persistent airflow obstruction after SARS-CoV-2 infection. Highlights:
SARS-CoV-2 and individual proteins may promote mitochondrial fragmentation (fission) leading to apoptosis (cell death) in airway epithelial cells (or any cell).
A regular human coronavirus, OC43, seems similarly hostile to mitochondria, though possibly to a lesser extent.
And, SARS-CoV-2 may sabotage the normal reflex (HPV) that shunts blood away from low-oxygen parts of the lung via the M or nsp9 proteins’ effects on the mitochondria of arterial smooth muscle cells. This impairs the normal mechanism for compensating for airflow reduction by keeping pulmonary blood-flow focused in oxygen-supplying regions and redirecting blood oxygen to where it is needed.

Background: The Kids Lungs’ Study
Although I lack a background in physiology or pathology, I try to supplement any observation of virus or vaccine-associated symptoms with some basic research on how the affected system is supposed to work naturally. In September, I posted a review of a study about kids’ lungs after reported infection with SARS-CoV-2, and dropped the ball on my background research.
The study in question, Heiss, et al., recruited kids self-reported as recovered from or still enduring symptoms (“Long Covid”) from the virus, and some not-reported-infected kids to serve as “controls” (really just showing what would normally have been expected in the scans in question).1 The kids were scanned with MRIs to measure airflow and blood-flow in the lung. The two shocking takeaways:
The lungs of the self-reported “recovered” kids were nearly as dysfunctional as the “Long Covid” kids. Presumably, the authors inadvertently introduced a bias for parent-observed, but unreported long-term effects in the “recovered” group in that these parents were more likely to volunteer for the study. But it is at least possible that these results are representative for all test-confirmed infected kids, which would be horrifying.
Circulation defects, as might be expected from micro-clotting, were not the only problem. There were air-flow defects as well, without any associated fibrosis to explain them.
The authors provide this triplet of representative example scans:
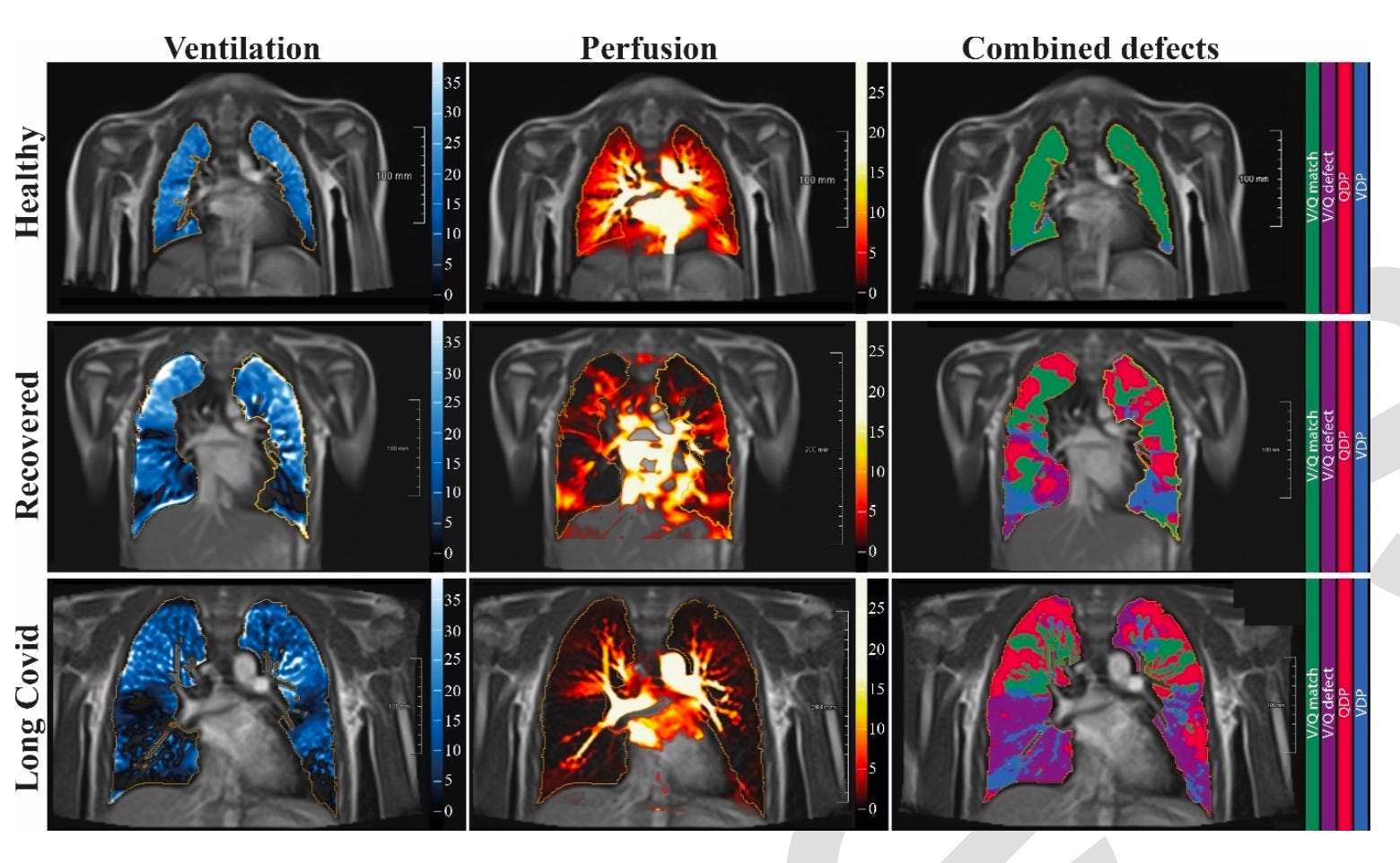
My linked post includes the full spread of scans and results. Not much differs from the examples above. The resulting rate of “blue” and “purple” detected by the study authors’ computers, absent fibrosis, is astonishing.

I commented, naively:
Lack of air-flow (ventilation, or V, in blue), outside of physical trauma, would typically suggest fibrosis or active inflammatory action (as I understand; pathology is my weak point). The authors did not find either. They did not perform additional tests that could shed light on the etiology for the widespread loss of ventilation function observed.
A new study clarifies the significance of these findings and provides a potential answer to the etiology.
Impairment of the normal hypoxia response
The subject of today’s post is Archer, et al.:2

This study addresses what I overlooked in my prior post: Hypoxic Pulmonary Vasoconstriction. Normally, pulmonary artery smooth muscle cells restrict blood from parts of the lung that do not have oxygen. As an example, if you hold your breath, the arteries in your lung constrict; this prolongs the function of the heart and brain by hoarding previously inhaled oxygen away from the lungs. Or if you have an infection clogging up your airways, blood is directed away from clogged portions, ensuring that pulmonary blood-flow only “draws from productive wells” and thus keeping arterial oxygen saturation from being diluted.
However, there are tradeoffs from long-term vasoconstriction and as such it can be considered pathological in some contexts, even if it is only a bystander reflex to an underlying airflow problem.
Without this reflex, blood continues to flow to parts of the lung without air. This can drive the so-called “V/Q Mismatch” which, as discussed in the previous post, helps diagnose what is causing shortness of breath or other respiratory symptoms. In other words, the Hypoxic Pulmonary Vasoconstriction reflex needs to be considered whenever there is blood not going to parts of the lung that have air or going to parts of the lung that don’t have air. The reflex, if functioning, should ensure there is only both air and blood or neither in any given location of the lung (except the very top and bottom).
Returning to the “mysterious” scans in Heiss, et al., the questions become clarified in light of the HPV reflex:
Why is the reflex failing in lots of air-flow obstructed regions?
Is this failure contributing to persistent air-flow obstruction?
Is the reflex, rather than clotting, driving defects in blood-flow?
Archer, et al., offers insights for the first two. The third is self-explanatory and doesn’t require much discussion.3
The authors do lots and lots of experiments and come to a number of conclusions. I spent much of yesterday reviewing the paper, but I won’t be burdening the reader with fine details. Essentially, I do not feel that the authors prove anything conclusively; but their theory is provocative in of itself. This post will only superfluously review the findings and theory offered regarding the three questions above. The theory is considered compelling in light of the observed airflow and blood-flow anomalies in Heiss, et al.: Here is something that might explain the mystery.
1: Why the HPV reflex might be failing.
The oxygen sensing mechanism that directs the Hypoxic Pulmonary Vasoconstriction reflex is still not perfectly defined.4 Among the candidate "O2 sensors" is the mitochondria of the arterial smooth muscle cells which do the constricting. Archer, et al. focuses on this mechanism, as recent work from their own lab has previously concluded that a particular protein complex in mitochondria (ETC Complex I, discussed below) is essential for HPV.5 These conclusions are still a matter of debate as there appear to be redundant mechanisms for O2 sensing; in vitro results may not reflect what happens in a living organism; etc.6 But Archer and co. are confident that mitochondria ETC Complex I is the O2 sensor that drives HPV.
So, they test whether infecting human pulmonary arterial smooth muscle cells with a lentivirus that codes for the SARS-CoV-2 nsp7, nsp9, or M proteins lessens the rise in intracellular calcium in response to low oxygen. As a control, they infect the same type of cells with a lentivirus coding for the GFP protein. The result:

They propose that the mechanism for this impairment is interaction with the mitochondrial electron transport chain gene pathways and depression of ETC Complex I, the authors’ posited “O2 sensor.” Infecting airway epithelial cells with SARS-CoV-2 down-regulated ETC Complex I genes (NDUFAB1, NDUFS7 and NDUFS8), among myriad other effects. In lung epithelial cells, transduction (lentivirus expression) with the same three SARS-CoV-2 proteins, and infection with regular old human coronavirus OC43, inhibited ETC Complex I activity as well.

They model their proposed theory of Hypoxic Pulmonary Vasoconstriction suppression in the graphical abstract / Fig. 8, seen above on the right. If virus or viral proteins are reaching the mitochondria of the smooth muscle cells around pulmonary arteries, they could be blinding the 02 sensors that should shunt blood away from areas of the lung with no oxygen. This exacerbates the shortage of oxygen in the body by allowing pulmonary blood-flow to “draw from unproductive wells” (lowering oxygen saturation), as well as allowing previously inhaled oxygen to go where it isn’t needed (diminishing the share of oxygen received by other organs at any given saturation).
2. Is this failure contributing to persistent air-flow obstruction?
In effect, it does not matter if the authors are correct about the exact mechanism for impairing HPV. (Unrelated to ETC Complex I but corroborative in a broader sense, HPV was also impaired when they infected mice with the MHV-1 coronavirus, indicating that this is a recurrent feature of coronavirus pneumonia.) Additionally, HPV failure could simply be a result of persistence of hypoxia; the O2 sensor may simply become desensitized after enough time passes. Whether it is via interaction with ETC Complex I or a different pathway that is responsible for regulating HPV, the results in Heiss, et al. demonstrate that impaired HPV may be a common and persistent pathology of infection with SARS-CoV-2.
Archer, et al., as it turns out, also indirectly propose an explanation for said persistence. Their model of hypoxemia exacerbation continues:

The arrows on the left correspond to their other findings regarding myriad ways SARS-CoV-2 promotes apoptosis via mitochondrial dysregulation (though, it’s arguable that these are innate immune responses designed to limit viral replication, and the authors mention the same possibility at multiple points). Nothing is groundbreaking here; it is already a given that the virus destroys airway epithelial cells and leads to some degree of pneumonia. What Archer contribute on this front is confirming that their three individual proteins of interest are just as destructive as the virus itself, showing direct mitochondrial toxicity rather than an immune-mediated response.
What they propose in the image on the right, however, is that the sabotage of the Hypoxic Pulmonary Vasoconstriction reflex also prevents stopping the “leak” in injured tissue: Continued blood-flow leads to continued leak of fluids and blood cells into areas that would normally be cut off by HPV. This supplements early findings by Caravita, et al. regarding severe Covid-19 patients, confirming apparent failure of HPV.7
The consequences of this mitochondriopathy include impaired HPV, which inappropriately floods capillaries in infected segments with blood [right], exacerbating capillary leak and promoting V/Q mismatch [blood-flow where there is no air]. Excessive apoptosis [left] damages the alveoli and contributes to [Diffuse alveolar damage]. The loss of HPV combined with AEC apoptosis exacerbate systemic hypoxemia in coronavirus pneumonia syndromes, including COVID-19 pneumonia.
This theory, besides explaining observations in severe cases, also seems compelling as an explanation for the persistent airflow obstruction in absence of fibrosis but presence of blood-flow in the post-infection kids’ lung scans by Heiss, et al. The virus may promote a lingering, self-reinforcing pneumonia without fibrosis. (Though here, again, my weakness in pathology and physiology prevents me from interpreting whether the images obtained by Heiss, et al. support persistent alveolar leak or not; my understanding is that MRI doesn’t really image pneumonia).
Naturally, there are also implications for Long Covid, where disruption of mitochondrial energy pathways are a prominent subject of attention.8
Archer, et al. finally propose that augmenting HPV (encouraging vasoconstriction in hypoxic regions of the lung) may be a new treatment strategy.9
Wrapping up, their findings that regular coronavirus induces many of the same pathologies once again suggests either that the results in Heiss, et al., may not be remotely representative of most post-infection outcomes; or perhaps, that long-term persistence of air-flow obstruction due to HPV impairment might be a common outcome from first encounter with coronavirus that simply goes unnoticed in childhood, and eventually heals.
If you derived value from this post, please drop a few coins in your fact-barista’s tip jar.


Heiss, R. et al. “Pulmonary Dysfunction after Pediatric COVID-19.” Radiology. 2022 Sep 20;221250.
Archer, SL. et al. “SARS-CoV-2 mitochondriopathy in COVID-19 pneumonia exacerbates hypoxemia.” Redox Biology, online ahead of press at sciencedirect.com.
Archer, et al. do not look at question 3. Their results on mitochondrial attack suggest, instead, a second means by which SARS-CoV-2 may destroy all sorts of infected cells besides replication and lysis; on the other hand, these “attacks” may in fact be innate immune responses by the same cells designed to limit replication.
However, it seems implicitly possible that the pathogenesis of SARS-CoV-2 Hypoxic Pulmonary Vasoconstriction interference could cut in both directions at once: It could impair HPV where it is needed, and perhaps, by interactions with proteins not investigated in this study, promote it where it is harmful.
Tarry, D. Powell, M. (2017.) “Hypoxic pulmonary vasoconstriction.” BJA Education, Volume 17, Issue 6, June 2017, Pages 208–213.
Dunham-Snary, KJ. et al. (2019.) “Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction.” Circ Res. 2019 Jun 7;124(12):1727-1746.
Dunham-Snary, KJ. Archer, SL. (2019.) “Response by Dunham-Snary and Archer to Letter Regarding Article, “Ndufs2, a Core Subunit of Mitochondrial Complex I, Is Essential for Acute Oxygen-Sensing and Hypoxic Pulmonary Vasoconstriction”.” Circ Res. 2019 Sep 27; 125(8): e35–e36.
Caravita, S. et al. (2020.) “Haemodynamic characteristics of COVID-19 patients with acute respiratory distress syndrome requiring mechanical ventilation. An invasive assessment using right heart catheterization.” Eur J Heart Fail. 2020 Dec;22(12):2228-2237.
From Archer’s commentary in theconversation.com
Our team’s infectious diseases expert, Gerald Evans, notes that this discovery also has the potential to help us understand Long COVID. “The predominant features of that condition — fatigue and neurologic dysfunction — could be due to the lingering effects of mitochondrial damage caused by SARS-CoV-2 infection,” he explains.
Not sure if it's related, but I am five weeks post-Covid (exposure on 9/25, first symptoms 9/27) and the cough is the weirdest part. The phlegm continues to this day, though slowly abating. I have not had a cold in years, but I have never had the drainage/clearance issues for so long. Otherwise I did not have a bad case and I rebounded after a week of work-from-home.
Kudos (not really) to the inventors of this virus. It is truly a unique experience.
I had covid last year and the defining physiologic feature to me was the amount of hypoxia vs lack of illness. I didn't really feel that bad and I even had a completely normal lung CT, but I could desat myself into the 80s within 30 sec of holding my breath. My conclusion at the time was that it must be either a shunting issue (as described in this study) or a red blood cell problem.