
Omicron Origins: Part 1
Why a lab-grown origin theory is necessary.

The seemingly random jumble of mutations in “Omicron” - BA.1 - suddenly becomes a set of markers for a two-part evolution when compared with BA.2, granting far more insight into the origin of late 2021’s mysterious sibling strains.
The Summary/Spoiler post for this essay provides a condensed overview, and additional comments on BA.4 and BA.5.
Part 1 of this essay, below, examines why a “wild” origin does not fit the story that comparing the two siblings reveals.
Part 2 discusses the siblings’ more likely development - and not accidental, but intentional - inside a lab.

Why Bother?
Last month’s treatise on BA.2, “Rashocron,”1 responded to the “Mystery of the Two Omicrons” by asking why we should accept that there is any mystery here at all. If the identity of a virus - its character and its genome, its “this-ness” - is a figment of our limited perception of reality, an imaginary particular applied to a horde of differences, why should be surprised if from time to time the figment suddenly no longer applies?
That question still holds - but here, we will be taking the opposite approach. We will attack the mystery on its own grounds, which is to say the grounds of genomic forensics, and solve it likewise.
That will, however, be the only time the “O” word is used to refer to the true subjects of our mystery: BA.2 and BA.1, neither one of which is more “Omicron” than the other, and whose departure from their root ancestor may be in reverse order of their discovery by human instruments (and everything in our analysis of BA.2 and BA.1 will apply to explaining any further siblings that reveal themselves).
Test Tube Babies
1: It Wasn’t Before Wuhan (unless B.1 was before Wuhan)
First, a review of why it is necessary that the origin of BA.2 and BA.1 took place in a lab (uninterested readers can skip to Part 2 for the origin theory reveal). Chronologically, the first alternate theory to dispense with is Ethical Skeptic’s “2+4 year ancestor theory,” or at least the potentially unfair simplification of it that I will recite here.2 The 2+4 year theory asserts that the last time BA.1 (and BA.2) and “the Wuhan strain” were the “same” virus was circa early 2018. BA.1 (and BA.2?)’s ancestor spread undetected through east and central Asia, India, the Pacific islands, and Africa, while Wuhan was “elsewhere.”
Both were accumulating almost two years’ worth of mutations up until the point of Wuhan’s official first sequencing - so when we inspect the difference between BA.1 and BA.2 and Wuhan, we have to bear in mind that some of the apparent “mutations” of BA.1 and BA.2 (about one-third) are actually pre-sequence Wuhan mutations, and the rest (about two-thirds) are post-branching BA.1 and BA.2 mutations. It’s a fun proposal, if a little over-reliant on confidence that evolution must obey a consistent schedule.
But as it turns out, BA.2 reduces the amount of mysteries the 2+4 year theory “solves” from 1 of 1 to 1 of 4. If BA.1 and BA.2 branched off from a 2018 pre-Wuhan ancestor, what would that look like in the 2+4 year theory view?

Note that synonymous mutations refer to those where a letter (nucleotide) changes, but the word (amino acid) remains the same (as some words have up to six possible spellings). These are mutations that ordinarily don’t respond as much to evolutionary pressure - the human immune system has no way to “ask” the virus to stop spelling words a certain way, for example, while it can “ask” for a new word by targeting the current word with T Cells and antibodies - but might still occur at slower or faster rates for other reasons, such as a result of a mutation to the bits of the virus that perform the job of copying and error-checking the code.
Now, this diagram of the 2+4 year theory is a bit of a brain-breaker, but that’s the point - to show the limits of the “molecular clock” in reducing the mystery. If the differences BA.1 and BA.2 have with the Wuhan sequence are 2/3’s alike and (on BA.1’s scale) 1/3 different, the dogma of the genetic clock would insist that they split around the same time that Wuhan emerged. Half of their “shared” differences with Wuhan are therefor Wuhan’s own pre-sequence mutations from the shared ancestor, and half are BA.0’s mutations. For whatever reason, both Wuhan and “BA.0” are both very pressured in direction of mutating in the receptor binding domain and the spike protein generally. BA.1 and BA.2 share only 5 synonymous variations from the Wuhan sequence, as opposed to their super-abundance of protein-altering nonsynonymous changes - again, a sign of high selection pressure in the era before their split.
But then there is the three-fold mystery of what happened after the split. First, a note on language. Whether BA.1 or BA.2 “broke” from BA.0, the opposite “sibling” essentially comes into existence at the same point. But it could also be the case that the opposite sibling remains in the same evolutionary context, while the “break” sibling enters itself into a new context. In this way, you could say that perhaps the “break” sibling is “older” than the opposite sibling, which does not experience a change to its evolutionary context until later on. (In fact, this is what will be proposed in Part 2). Resort to such accidents of timing could be used to try to rescue the 2+4 year theory. Perhaps the spike pressure on BA.0 slows down, BA.2 “breaks” into a new context and experiences a pick-up in evolutionary pressure (now on “open reading frame 1”), and later BA.1 either breaks from 0 again or the pressure on 0 suddenly radically alters - in either case there’s wiggle room. But using wiggle room to “rescue” the evolutionary clock in fact demonstrates that the evolutionary clock is ridiculous nonsense to begin with. The rate of evolution can radically change, when genes enter a context of high evolutionary pressure. BA.0 was under apparent high pressure on the RBD of the spike protein; BA.1 shows signs of radical pressure (in favor of deletions and insertions) on the NTD of the spike protein, and BA.2 shows signs of permissive high pressure (both synonymous and nonsynonymous changes favored, as if to mutate for its own sake is the goal) on the genes in the “Orf1” set of polyproteins (whereas BA.1’s changes here are proportionally less conspicuous, though not non-zero). Both siblings look “cooked,” and both look like they were cooked the same way for a time, and then separately cooked two new ways. As I have yet to succeed in my attempt to rebuild the mutation list on my computer several times, I have given up and simply stolen and annotated Andrew Rambaut’s excellent but temporally backward mutation list. Reading the following mutations in reverse, the fact that BA.0, BA.1, and BA.2 all experienced pressure in different “directions” should appear obvious:
So there is in fact no need to back-date BA.1 and BA.2 to “before Wuhan,” or to attribute 1/3 of their changes to Wuhan, in order to justify the large overall count of mutations. When either is looked at “vs Wuhan” it may seem like they have spent their voyage to discovery “wandering” through random mutations; when either is looked at vs the other, it is clear that their voyage, whenever it started, was one of universal and yet variable high pressure. If that is the case, then we can safely work with the mainstream-aligned premise of a genetic origin circa Wuhan or later.
So the 2+4 year theory is thus forced; it doesn’t actually offer a lot of answers; and in fact it renders one of the mysteries less well-answered than before. Namely, it implies that the “break” between BA.1 and BA.2 was two to three years in the past. In other words, these two siblings are as different from each other as they were from the Wuhan strain to begin with. And yet neither gained whatever magic mutation allegedly shifts them from undetected infection and (high-pressure) evolution in the intervening time, and both coincidentally evolved that mutation circa October or November of last year? Certainly it’s exciting to imagine some type of merger, a co-infection-spawned genetic marriage between wandering invisible viruses (which nonetheless fails to remove two to three years of difference) that renders them detectable, perhaps even a three-way with Delta that donates the triplet of D614G genes.
And, despite the clear changes in evolutionary pressure experienced by BA.0, BA.1, and BA.2, it’s obviously possible that some of the “shared” mutations we are attributing to BA.0 (and pre-sequence Wuhan) could have coincidentally been co-evolved by BA.1 and BA.2 separately. The BA.0 RBD signature is our arrogant inference of the pre-break pressure environment, but doesn’t dictate the actual order of mutations that occurred. Both BA.1 and BA.2 have some (3 and 4) unique edits to the RBD, which could suggest a period where both were still in the RBD pressure environment post-“break,” or a reentry into that environment shortly before they were sequenced (but while they were still separate viruses). But even if they evolved the same pivotal mutation - let’s say N501Y - separately after re-entering an environment of pressure on the RBD, supposing that they did so at the same time - as in, not by accident at any time in the intervening years, resulting in earlier detection - is a stretch. Sure, it fits in the “Rashocron” model of total uncertainty; but it doesn’t satisfy the rules of the mystery.
Ultimately, the 2+4 theory, if playing by those rules, leads to the assumption that BA.1 and BA.2 spent ~30 months in an environment when a 1 per 30 month mutation was all it took to render them visible to human detection, and both didn’t “roll” that mutation until the 30th month. And, both were invisibly spreading undetected for a total of 48 months, the first 18 of which were in a high-RBD pressure environment - which, again, reduces the need to refer to the genetic clock to begin with, but also implies that there should already be antibodies against this thing everywhere.
Of course, there are still other ways to rescue the theory; in fact it really takes a while to debunk them all. For example, one could propose that there was no co-mutation, but instead that what we have named “BA.1” acquires the “detection mutation,” and reveals pre-existing BA.2 wherever it spreads. In other words, BA.2 becomes detectable when BA.1 is geographically prevalent, due to hypothetical synergistic properties. But, once again, both were everywhere all along (and perhaps the pre-detectable version of BA.1 is also synergistically revealing itself, but being mistaken for a novel sub-strain). There is a glaring problem with this one: Why isn’t BA.2 being “revealed” in the US; and why so rare in the UK and Germany?

It seems more likely that both are, in fact, “novel,” either in their presence on Earth or in their co-mutation of whatever makes them detectable (if they weren’t before). So the stupendously unlikely co-mutation problem remains. (Note that I still find Ethical Skeptic’s theory on an early release compelling; I just don’t think it has anything to do with the origins of these two sibling strains).
2: It Wasn’t After Wuhan
To reduce the implausibility of the “coincidental co-mutation” that explains BA.1 and BA.2’s simultaneous discovery, as well as reduce the implausibility of such a long period of non-detection, we need to move the “BA.0” split to after Wuhan. Here, the “chronic infection” theory almost gets in the door. The problem is that this thing did not branch off from Wuhan “after” anything. If someone was long-term-infected with Delta, BA.0 must have magically un-mutated all of Delta’s differences from Wuhan.
Likewise, if someone was long-term infected with Alpha - with which BA.1 shares some similar mutations, but not so much BA.2 - “AIDS patient BA.0” must have un-mutated the synonymous Alpha mutations C241U, C913U, C5983U, C14676U, C15279U, U16176C, while only keeping C3037U, and having gone on to only make 4 additional synonymous mutations (vs Wuhan) at the same time, and also favoring U>C mutations less than post-Wuhan strains have done (indicating a non-human-infection signature). And then, “AIDS patient BA.2” must have gone on to magically undelete 9 Alpha-deleted letters in the NTD. We are already well past the bounds of implausibility with this one. It’s far more likely that, because BA.1 and BA.2 are collectively throwing 85 mutations (only 38 shared) at the Wuhan genome, they’ve merely repeated some of the real-world mutations either by accident or because those edits remained favorable even in the altered, likely non-human pressure environments both experienced.
In fact, it’s questionable whether we should grant that BA.0 branched off from a post-Wuhan, real-world copy of SARS-CoV-2 at all. The most conspicuous post-Wuhan mutations that BA.0 still bears are a triplet of mutations that were “developed” in the first months of 2020, and that, when combined together, transformed the Wuhan strain itself from something that was a bit “hard to detect,” in that it seemed like a product of TV news hype, into a world-shut-downer (which was still a result of TV news hype, but at least now there were “full” hospital wards somewhere, as though that wasn’t already the case before). These mutations appeared separately in sequences throughout the world, but even the incredibly successful D614G mutation didn’t seem to make much of a difference until it was combined with the other two, as first appeared in a sequence in China in January and in Italy in February 20. Like D614G, the other two mutations in the triplet spontaneously appeared in sequences around the world where the “Wuhan” strain already had feet on the ground in late February.3
This triplet includes not only D614G but “Orf1 P4715L” (or “Orf1b P314L,” depending on the numbering scheme used), a rather dramatic amino-acid swap to the virus’s otherwise “caution: fragile” code for its own gene-copying protein, and, interestingly, a synonymous mutation at letter 3037, from C to U (again, synonymous because the amino acid spelled by the three-letter set that contained the “C” did not change). We can thus conclude that C3037U, despite not changing an amino acid, is somehow critical to the overall fitness benefit conferred by the triplet, or that it is a signature belonging to an undetected “Cinderella” version of the original Wuhan virus - one that would be present even in January, 2020 isolates, but would be dismissed as a quality error if it happened to come up in a sequencing of those isolates.
This proposal does not place us too far in the wilds, where we might flaunt the rules of the mystery. If the Wuhan virus was grown in a lab, it would potentially “self-attenuate” due to successive generations spent infecting cells that are not protected by host immune systems. This attenuation would self-reverse, if not immediately (as in during the course of a single confrontation with the human immune system), then likely within a few transmissions - for it would turn out to be the case that the high-fitness version of the virus is not lost in the lab, but still resides somewhere in the “swarm” in which the low-fitness version has come to dominate.
Here, however, note that I do not buy Sirotkin’s suggestion that such a reemergence takes months or years or decades.4 What we are dealing with here is a competition between two ultra-powerful gravitational forces in the world of asexual genomes: Natural selection, and clonal interference. In the former, contextually high-fitness versions of the genome, because they replicate more successfully (the character by which fitness is defined), become more prevalent; in the latter, a good-enough version of the genome will still outcompete more fit versions if it already has the advantage of arbitrarily high-coverage in the context of “all locally existing versions of the genome” - say 99.99%. If underlying circumstances suddenly render “good enough” “sub-par,” the inertia of clonal interference will not protect the no-longer-fit version for long. Natural selection is now already the dominate force in this case; so we can expect its effects to become visible sooner, rather than later.
For example, the intentionally attenuated, live Sabin polio vaccine demonstrates that RNA viruses can recover their normal level of fitness within a single infection. The versions of the “vaccine” that were pooped out by the kids who received it could in fact cause paralysis, if transmitted to someone who hadn’t also been vaccinated. Further, around a dozen recipients per year still developed paralysis. A few passages through actual, living cells are all that are required for natural selection to undo clonal interference, when a virus is facing real immune systems. Meanwhile, the .01% version remains invisible to sequencing until it has driven the “good enough” version into the swarm’s periphery - where it will likely be banished into the dustbin of genetic history before long. (Perhaps the reader is beginning to see how this tangential visit to the past is instrumental to how we will be thinking about BA.1 and BA.2’s staggered “discovery,” in the context of a released, lab-grown strain.)
Release from the lab thus not only placed SARS-CoV-2 in a context where new mutations would quickly rise to the top, but where deleterious mutations that had already risen to the top would reverse themselves. In fact, neither event seems to have happened as much as expected - suggesting, in other words, that the lab did a pretty good job of keeping the original version in high fitness form.5
The P4715L change is “radical” in that it dispenses with “rigid” Proline in favor of more vanilla Leucine. In an early analysis, this seemed to result in a higher rate of mutation for triplet-bearing versions of the virus;6 though that higher mutation rate could itself be an artifact of better fitness. But, running with the suggestion, we could imagine that in a pre-release “lab” version of the Wuhan strain, insertion of Proline led to more stable replication performance in the gene-copying RdRp protein built by the corresponding genes, and this led to the dominance of this pre-Wuhan variant in the environment of lab passage; it’s just one hypothetical among many which could apply. In this scenario, the more mutagenic, ancestral L-bearing version rises to the top after two or three successive infections in the real world. If the reader buys this timeline, then perhaps Sirotkin’s theory that “live viral vaccine deattentuation” can still be a threat months and months after release, if not forever, will seem less plausible.
At all events, the synonymous C3037U mutation becomes part of the signature of this pre-Wuhan Cinderella version of the virus - the version that disagrees with the Wuhan sequence on both P4715L and D614G, and went on to be deemed “B.1,” whereas D614G alone in fact died out. B.1 represents the ancestor, “reverse G614D” was only an inconsequential signature belonging to the Wuhan sequence, and everything was about Wuhan’s L/P defect in the all-important gene-copier protein. The triplet’s simultaneous emergence in disparate corners of the globe naturally suggests that these “mutations” were in B.1 from the start, and that B.1 was already present in the original release, as well as in any pre-B.1 samples that might be ordered online and serial-passaged through a mouse by a researcher to this day.7
What’s more, neither BA.1 nor BA.2’s spike proteins bear any of the common non-B.1 mutations from early 2020. Here, for example, are the “top 80” spike protein mutations in sequences up to May, 2020 (D614G+ merely refers to mutations that are not seen except alongside B.1, red are “high frequency”):
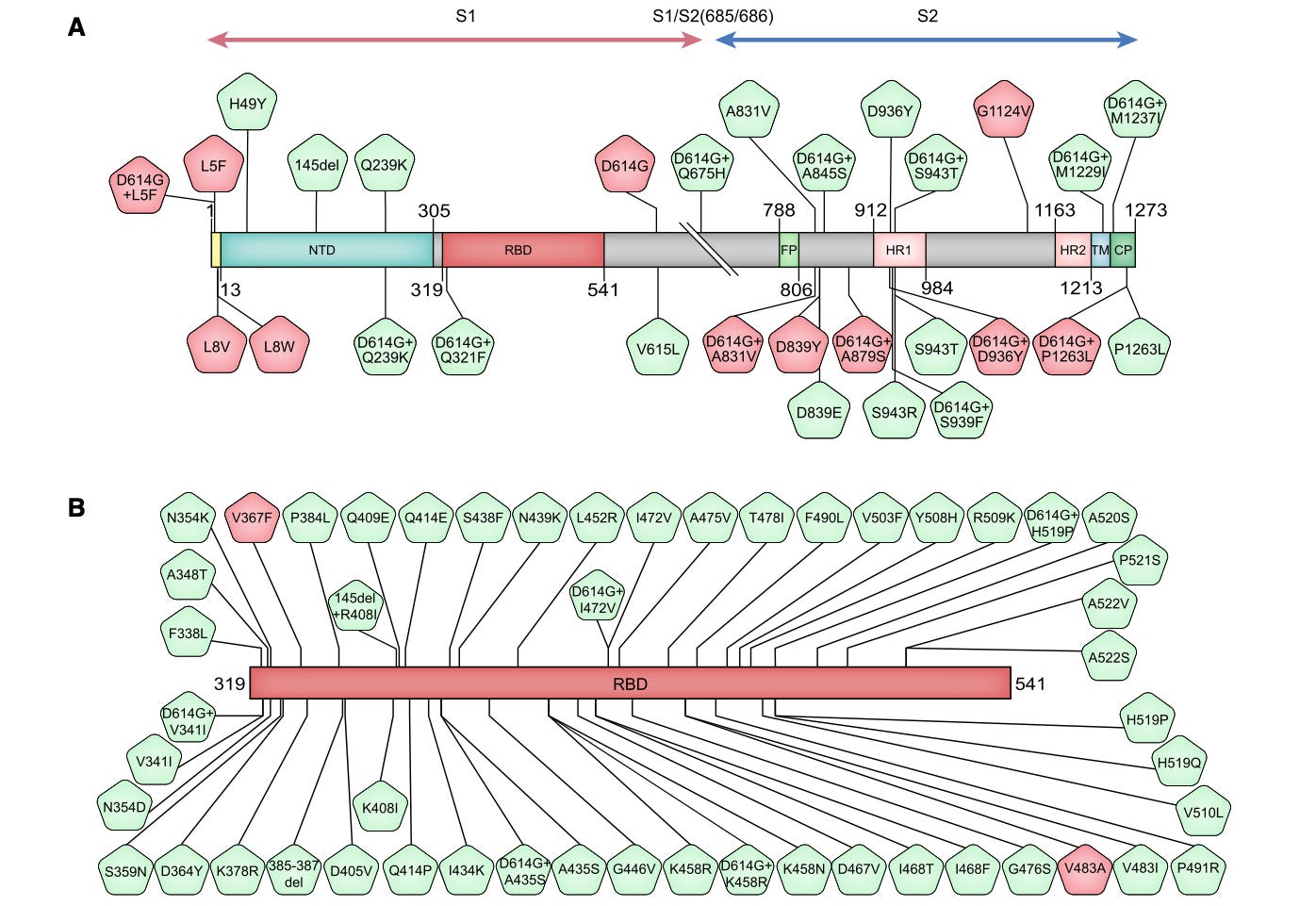
None of these are hits for BA.1 or BA.2, except for the deletion at 145, which is only on BA.1, and is part of BA.1’s post BA.0-era NTD mutation signature. (Nor are they in Alpha or Beta, either; someone should probably look into that.) This doesn’t prove that they could not have descended from a post-Wuhan sample - obviously, there would have been billions of versions of post-release Wuhan and B.1 that did not bear these specific mutations out in the wild; and the threshold for “high frequency” was only .1% or greater. But, it’s conspicuous that there is no trace of the early 2020 real-world spike mutation signature in a product that otherwise traces… to “early 2020.” It seems just as plausible that BA.1 and BA.2 descended from a Wuhan sample that either had B.1 already in it - or that, naturally, it was edited on purpose to include the vaunted B.1 triplet (and the same goes for Alpha and Beta, as well; someone should probably look into that).
BA.1 and BA.2 did not branch off of the human-spreading virus before Wuhan; and it seems likely that they did not branch off after Wuhan. They descend from that original version. Resemblance with B.1 and some of the other variants is the product of B.1 already existing in Wuhan samples, re-entry into conditions that favored the same mutations to begin with, coincidence, or intentional edits.
So, we have tried to fix the biggest “problem” of the 2+4 theory, only to find that we are still left with most of it: 23 months of non-detection, instead of 48, and something like 8 months of coincidental non-co-mutation of the magic “1 per 23 month detection mutation” followed by coincidental mutation, depending on where we place the split between BA.1 and BA.2 on this model (which could really be anywhere, since we have thrown out the genetic clock).
Let’s put BA.1 and BA.2’s origins where they have obviously belonged from the start: in the lab.
Continued in Part 2.
See “China’s CCP Concealed SARS-CoV-2 Presence in China as Far Back as March 2018,” which was originally posted shortly before “Omicron”s (BA.1’s) arrival, and then retconned to incorporate the new bug. I haven’t yet re-visited to see if there has been an attempt to somehow fit BA.2 into the theory.
See Eskier, D. et al. (2020.) “RdRp mutations are associated with SARS-CoV-2 genome evolution.”
In the following quote, the occasional reference to a fourth mutation seems to be accidental (counting 23403 / D614G twice).
On the other hand, it is intriguing that between the first appearance of three co-mutations (on 14408, 23403, 3037) on January 24 in a Chinese isolate (EPI_ISL_422425) [this one was likely suppressed before spreading, but who knows] and their second co-appearance on February 20 in an Italian isolate (EPI_ISL_412973), there are at least six and possibly eight different viral genomes where three of the four co-mutations exist, with the exception of 14408C>T: on January 28 in Germany (EPI_ISL_406862), on February 5, 6, 7 and 8 in China (EPI_ISL_429080, EPI_ISL_429081, EPI_ISL_416334, EPI_ISL_412982, and EPI_ISL_429089); and two more Chinese viral sequences that failed our quality control standards and were eliminated from the overall analysis. Despite weeks of existence, 23403A>G [D614G] became the dominant form only after the appearance of the first Italian case with all four [three] mutations on February 20. If this form of SARS-CoV-2 is really more transmissible, the next question that needs to be answered is whether it is due to any one of the three mutations alone, or whether a combination of two or three are needed. Based on the lack of successful spreading of the virus in its absence and our results showing increased mutability in its presence, we speculate that 14408C>T could be cooperating with the other two mutations. Alternatively, altered mutation rate may be a byproduct and the RdRp mutations may act through speeding up or slowing down the replication process, which would in turn affect the viral load and virulence.
(There’s more to quote from this, but I need to get back to writing part 2.)
See Sirotkin, Dan. “A Grin Without a Cat.” (2021, December 3.) Harvard to the Big House.
This “human-optimized from the start” trait is part of the evidence for the Lab Leak™, as if the zoonotic origin theory ever actually required debunking - but it still remains an interesting question why the virus has been not very responsive to real-world selection pressure overall.
(Eskier, D. et al.)
See “Mouse Party,” which is also semi-required reading for Part 2.
Was there not reporting that CRISPR could impact the biological process of which traits pass on in mosquito's -- used to control eye color and malaria spread I believe. They were able to have a recessive trait (normally) overtake dominant traits with something like 100% success rate.
Could you not do the same with a virus?
Could you build a virus with mutating variables on the outside // high mutation // to ensure my "packaging" continues to grey man the immune system. Then a low mutation core to ensure delivery of specific outcomes.
The Chimera was the beast desired, Frankenstein may be the beast we got.
I don't grok obfuscation haiku well enough to understand TES to disagree with him ;-) He reminds me of someone who talks a certain way so you cannot disagree with him. Almost post modernist sokal hoax? Dunno. Maybe I am 2 orders of magnitude dumber than I think instead of only 1. I am honestly amazed at how you appear to have taught yourself immunology and genetics in the space of a couple of months, and yet have injuries from labouring, which makes me think you're a brickie :D
You are making this far more understandable than anything I have ever tried to read from his hand. Appreciate the effort.