


Omicron Origins: Summary / Spoiler
The Omicron siblings tell the story of their creation in their genes.

“Omicron” turned out to be a family of two novel super-mutant models of SARS-CoV-2, one of which came with further alternate versions. This is unlikely to have resulted from real-world evolution of any type.

Background
In February, I published my report on the origins of the Omicron siblings, relying on my rudimentary, caveman-like phylogenetic forensics to crack the case: Genes too different. Lab make thing.

As is my wont, I chose a plain prose format, without an outline.1 A complicated story was being told. A literal, real-world mystery - in which the best way to reveal the answer is to chase all the wrong leads first.
Since then, an alternate history of Omicron’s emergence has taken hold, forming a collective understanding which obscures the very same genetic evidence I used for my essay. I was particularly made aware of this in the comments for Modern Discontent’s recent overview of BA.4 and BA.5, in which it seemed as if I was the only one who remembered that 4 and 5 were sequenced so early and so geographically close to Omicron ground zero that it should be assumed they are not real-world descendants of BA.2 suddenly attacking from out of nowhere, but instead were already circulating from the start (in November).
And so, as a reference document for how this false memory of the Omicron era differs from the real events, I am now presenting the bulleted summary for my origins essay, giant text galore, which probably should have been included in the first version.
This summary will include comments on BA.4 and 5 that are not included in the original post. LCA (Last Common Ancestor) will be the only difficult but also essential acronym.2 For other unclear terminology, see the more thorough original post above.
-Not grown in “nature,” whether on a 2 year or 6 year timeline.
Defining “BA.0”
BA.1 and BA.2 have a lot of overlapping mutations, and a lot of diverging ones. This implies that at some point they were one virus, and then they split.
The virus that existed immediately before one of the siblings breaks away can be thought of as a historic “BA.0” ancestor, a long-lost version that led to both of the bugs that were finally discovered and sequenced in late 2021.
BA.0’s genome can be defined as “almost every gene that is the same in BA.1 and BA.2.” Some of these might be instances of convergent evolution, but since only .5% of the virus’s genes were changed vs Wuhan (depending on your math), mutations after the split between 1 and 2 were unlikely to be to the same gene.
B.1, (or B.1.1), not Wuhan, pre-Wuhan, or post-B.1, was the LCA between Wuhan-descendent SARS-CoV-2 and BA.0.
By the same logic as the previous bullet, it is super unlikely3 that the B.1 mutation signature (minus C241U) arose in BA.0 by random chance.
For BA.0 to have all three of Orf1 C3037U (a synonymous mutation, which does not change the amino acid inserted in the polyprotein), Orf1 P4715L (sometimes described as Orf1b P314L), and Spike D614G, and the same underlying nucleotide changes leading to the former two, makes it safe to assume that B.1 or B.1.1 (+Nucleocapsid RG203KR) is the LCA of BA.0.
Conversely, BA.0 is unlikely to have derived from any late-2020 circulating strains (the greek-letter variants), as to do so would have required coincidentally reversing numerous mutations, including synonymous mutations - despite BA.0 generally favoring nonsynonymous mutations, which should mean that any synonymous mutations in the LCA would still be visible, like B.1’s C3037U. As a counter-argument, BA.0 seems to have dropped B.1’s C241U.
B.1 may be a predecessor to the first sequenced Wuhan variant
*edit: This section is provisionally redacted, pending a new analysis incorporating B.1’s appearance in October, 2019 in Italy (after Wuhan-like sequences in the same samples) (Amendola, et al.). B.1 may have been circulating before the first reported Wuhan sequence, but still was essentially a genetic descendent of it. The text of this segment is left here in strike-through in case its removal renders any of the other segments unclear.
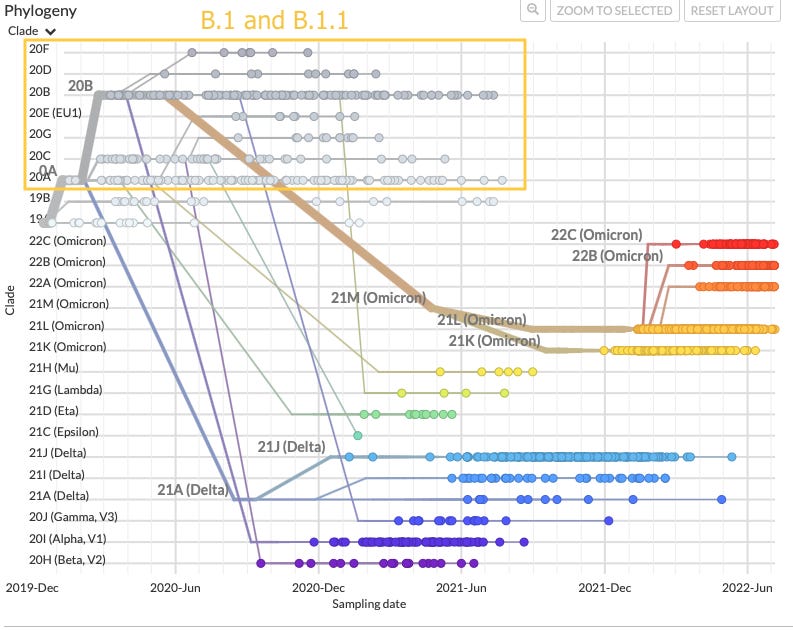
Obviously,somethingpreceded the Wuhan isolate / sequence. First in the sense that the SARS-CoV-2 genome was derived from some unknown, SARS-1-like source genome; and second in that human-to-human transmission between release and sequencing may have introduced unknown post-release mutations. So it is at least possible that the predecessor virus was in fact better at transmissionorstable lab performance / culturing than the virus that was detected and sequenced in Wuhan, and that spread semi-inefficiently before February.D614G was detected in multiple regions early on, including in Italy in February, and was associated with increased virulence (see original post above for citations etc.).If D614G alone improves fitness, it is curious that the other three mutations of the B.1 signature became fixed (the term for when one allele displaces all variant forms within an asexual genome).Overall, vs. Wuhanevolvinginto B.1, an equally-coherent or more-coherent explanation for this outcome is that B.1 was a separate release of either an improved Wuhan descendent or a predecessor to Wuhan which was attenuated by Wuhan’s mutations to Orf1.This theory deserves better examination than I give it here. Unfortunately, the history of B.1’s dispersed emergence seems to exist only in non-user-friendly raw GISAID data. A crude examination of this data is presented in the footnotes.4 At all events, what matters is that B.1 / B.1.1 is the LCA for the Omicron siblingsandthe 2021 VOCs.
And, B.1 / B.1.1 and the Greek letter variants don’t evolve much in real-world transmission
All Greek-named, late 2020-emerging variants of concern are B.1-descendent, but do not have any other known circulating predecessors. They also feature more mutations than the contemporarily circulating Wuhan or B.1-derived strains.
Again, vs. Wuhan evolving into B.1 and then Alpha, Beta, etc., an equally coherent or more coherent explanation is that the late 2020 VOCs were separately developed from B.1.1 in non-wild conditions and then released.
Conversely, vs. the Covid vaccine trials somehow prompting rapid mutation from circulating SARS-CoV-2 into the late 2020 VOCs, it is unclear why widespread distribution of the vaccines did not accelerate de novo formation and discovery of similarly “out of the blue” variants. Rather, now-wild versions of the VOCs (primarily Delta) went on to mutate at the same slow pace as the original Wuhan and B.1 in real-world transmission.
Changing evolutionary pressure over time
BA.0 had (a few) more mutations vs the “Wuhan” version of SARS-CoV-2 than the last circulating Delta strains.
But, both siblings further added many more mutations to BA.0. By so doing, they created a forensic trail that retroactively illuminates the path of development preceding their detection.
And so:
BA.0 mutations happened before either BA.1 or BA.2 (unique) mutations.
BA.0, BA.1, and BA.2 mutations all show a focus on different parts of the virus’s genes. BA.0, BA.1, and BA.2 all seem to have been subject to different types of evolutionary pressure.
Since synonymous mutations are either scarce or conspicuously grouped, this variable evolutionary pressure seems to have been extraordinarily high at all points along the way.
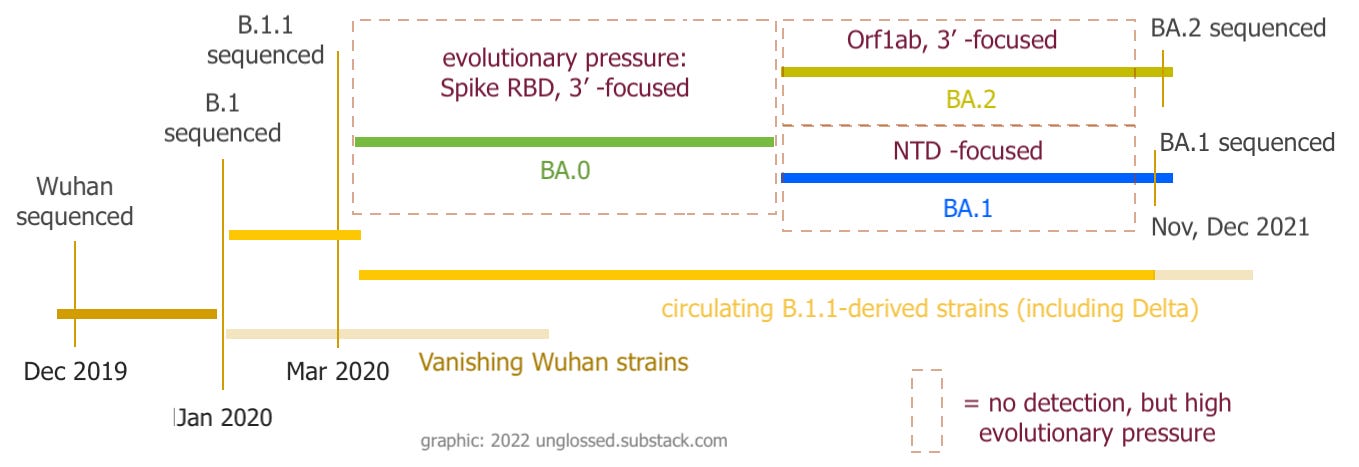
The raw mutation list, with B.1.1 mutations taken as part of the original template, tells the same story. BA.0, BA.1, and BA.2 all show a “focus” on mutating different regions of the genome, and thus were all subject to different evolutionary pressures.
Omicron as a “pre-Wuhan” variant doesn’t work
The blogger who goes by “Ethical Skeptic” proposed that the reason “Omicron” appears to have ~6 years’ worth of mutations is that 4 of those were between 2018 and 2020, and were distributed between the eventual Wuhan sequence and the eventual Omicron sequence.5 Some 2/3 of Omicron’s apparent mutations would therefor be split 50/50 between pre-sequence Wuhan mutations and contemporary Omicron mutations.
This math became strained as soon as BA.2 was revealed to have such a radically different sequence.6
More importantly, Ethical Skeptic’s account would propose that BA.0, and later BA.1 and BA.2 both spent 4 years transmitting and evolving undetected, and yet the two siblings simultaneously acquired some undefined mutation at the same time (after roughly 2 years post-division) that rendered them detectable, leading to their nearly simultaneous sequencing in South Africa.
One could instead resort to some theory that BA.1 acquired a mutation which synergistically rendered BA.2 more detectable. In this scenario, the suddenly-detectable new version of BA.1 spreads around the globe and “illuminates” previously undetectable BA.2 wherever it goes. But instead, BA.1 spread to most regions without revealing any pre-existing BA.2 at first. This neatly refutes the idea that BA.2 was in any sense “out there all along”:

from Yamasoba, D. et al.

The problem of simultaneous discovery applies to any other timeline for “natural” emergence.
Undetected community transmission, immunocompromised chronic infection, and animal transmission, are all weaker at explaining how the Omicron siblings were “cloistered” from humanity at large during their extensive evolutionary development from a pristine B.1.1 template.
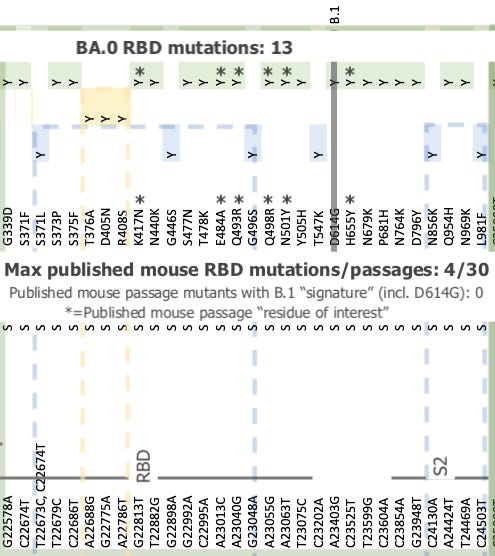
Absent effective cloistering, a scenario must again be imagined where a mutation was required to render both siblings detectable and transmissible in humans again, and that this “once in every 18 months” mutation somehow was achieved by both siblings simultaneously. And so animal transmission is particularly discredited here. Meanwhile, the mutation signature for BA.0, particularly in the RBD, matches mice rather than humans (see Part 2 for more).
A lab origin for BA.1 and BA.2, where no simultaneous mutation for human detection is required, but human detection is instead prevented because there is no real-world human transmission, better explains this outcome.
-Not released from a published mouse serial-passage study.
Many published studies attempted to adapt Wuhan-matching isolates of SARS-CoV-2 for efficient transmission in mice in 2020 and 2021.
Including from Baric’s lab. This was reviewed in Mouse Party.
However, none of these “resurrected” the B.1 signature, despite several dozen passages in some cases.
Since BA.0 contains the B.1 signature, it originated from B.1, not from a Wuhan isolate, and therefor not from any of the published mouse serial passage studies.
Nonetheless, BA.0 contains the brunt of the Receptor Binding Domain-located “mouse-like” mutation signatures that were initially attributed to “Omicron.” It also exceeds those signatures, suggesting that BA.0 far exceeded any published mouse serial passage experiment, perhaps by combining serial passage with immunization against mutations as they arose.
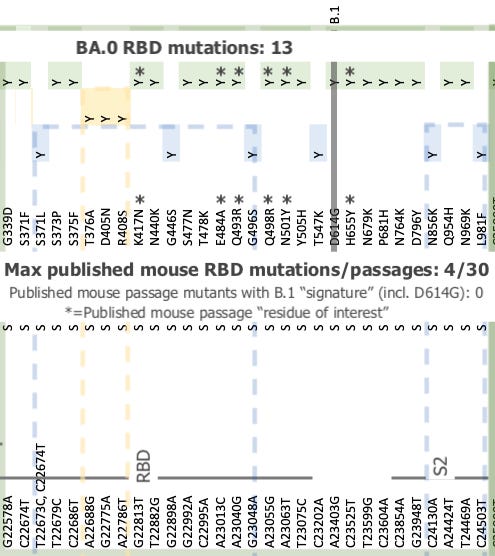
Background image shamelessly stolen from Andrew Rambaut. Published mouse passage studies / mutations compiled in “Mouse Party.” Further discussion in Part 2 of Omicron Origins. Thus, even before the split between BA.1 and 2, BA.0 has ventured beyond the “known universe” of mouse-directed Spike Receptor Binding Domain mutations.
Furthermore, post-BA.0-mutations indicated a shift away from the Receptor Binding Domain focus of BA.0, toward Orf1 (for BA.2) and the Spike N-Terminal Domain (for BA.1), suggesting that neither sibling’s evolutionary pressure derived from mouse serial passage after the split. Some other type of evolutionary selection or genetic manipulation was at play.
BA.1’s N-Terminal Domain also features the most radical mutations that were initially credited to “Omicron,” including the EPE insert. This could be a signature for lab recombination, as a speculation. In either case synonymous mutations remain uncommon, suggesting that long-term undetected natural transmission is not the culprit for these changes. Rather, BA.1 and BA.2 both add their unique finishing touches in a mad evolutionary rush.
Many possibilities exist for a lab origin scenario which could account for the mysterious changes in evolutionary pressure between BA.0, 1, and 2. A “lab loss + recovery” theory is offered in Part 2 of Omicron Origins, as a template.


-BA.4 and 5 did not emerge from natural transmission, either.
Sequenced too soon.
BA.4 and BA.5, which are really sub-variants of the BA.2 model, were both first sequenced in South Africa in January and February of 2022.7
Both feature “mutations” that would have to be described as “reversions” to Wuhan vs. BA.2, if supposing that they derived from BA.2 as initially sequenced.
However, it is implausible that the both coincidentally derived from real-world transmission of (post-sequenced) BA.2 in South Africa, but no other reversions worthy of a new “BA.X” designation have arisen anywhere else in the world, despite higher absolute levels of transmission outside of South Africa (the Delta-BA.1 recombinations are precedent for a different type of “alternate Omicron”).
It is far more plausible that BA.4 and 5, like BA.2, were in South Africa at the same time as BA.1 was first detected, but merely took longer to be caught on a sequence.
In the model where BA.4 and 5 are in the original release, BA.2 mutations that 4 and 5 do not share were acquired before BA.2 was sequenced. BA.2, 4 and 5 would share a “BA.2 family beta” LCA that preceded BA.2’s sequencing, with BA.2’s unique mutations vs. 4 and 5 being added after the division from this LCA. Thus, only Q493 has to be considered as a reversion (or last-minute convergent evolution between BA.1 and BA.2), while Orf1 L3201 and Orf6 D6 are preserved in 4/5 because the mutations in these genes occurred in BA.2 alone.
The spread of each Omicron sibling must also be interpreted in the context of in-clade competition. BA.4 and 5 are in the initial release, but only become more fit after BA.2 has generated negative selection against itself via immune response. More comments are provided in the discussion at Modern Discontent’s post. A follow-up post here will drive the point home that SARS-CoV-2 is not actually “evolving faster.”
If you derived value from this post, please drop a few coins in your fact-barista’s tip jar.


My goal is not to dictate conclusions to the reader, but to show the path which I believe leads to those conclusions. Otherwise, Unglossed merely becomes one more source for menu items in an infinite buffet of “available beliefs” that internet consumers can select from to fashion their own version of reality.
It just sounds too cool to not use.
Science term.
In Nextstrain, scatter view allows one to trawl the earliest “20” clade (B.1) and “19” clade sequences. However, the neat grouping into clades elides the degree to which B.1 and B.1.1 seem to parachute into different regions with no other consistently shared mutations. (Also, some 19 clade sequences after May are actually B.1.1).

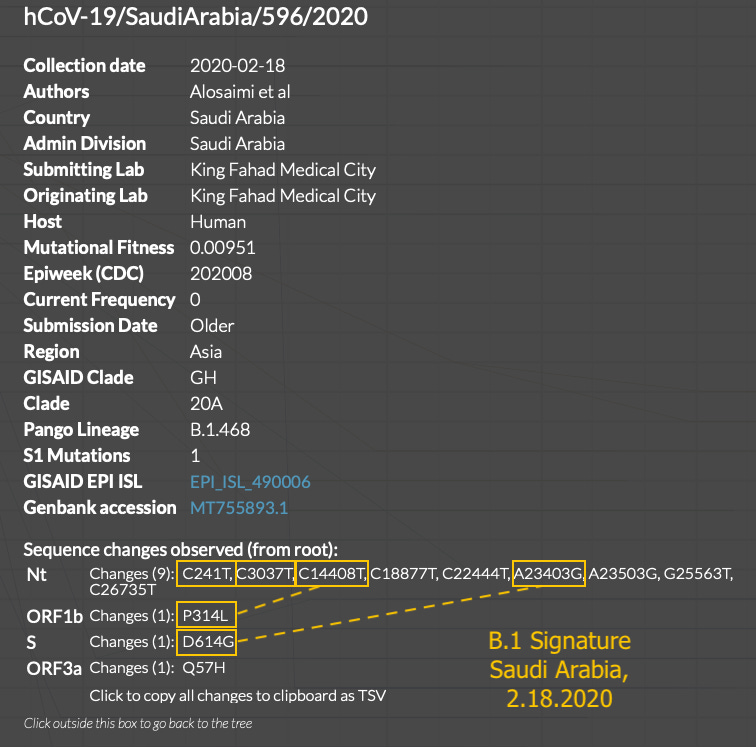


Weeks later, even though B.1 is still going strong, the Nucleocapsid protein, two-residue-spanning GGG>AAC change in B.1.1 appears, in both the northern and southern hemispheres at once:
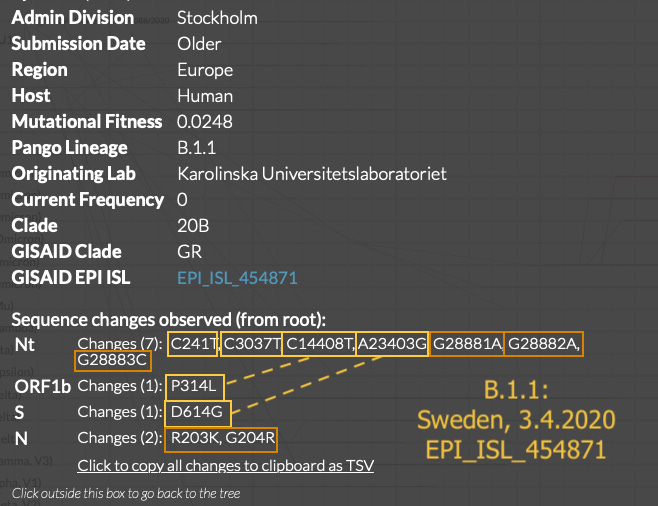
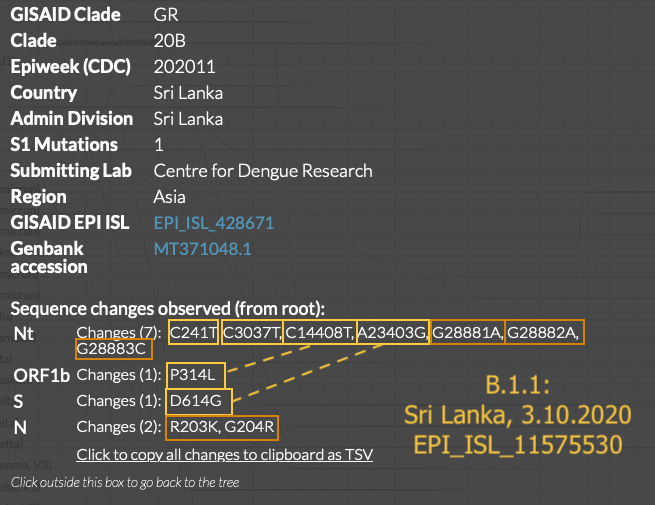

What is additionally notable is that the early 3.1.1 sequences are just as “clean,” if not more-so, than the early 3.1 sequences. The Morocco, Sweden, and Sri Lanka sequences are as unadulterated representations of B.1 and B.1.1 as it gets. Aside from the Colorado sample, early B.1 and B.1.1 do not show as much “mutational noise” as some of the contemporary true-Wuhan-derived sequences preserved in nextstrain.

Obviously, it may be the case that C241U, C3037U, and C14408U (P4715/314L) were merely “passenger” mutations on D614G when it first occurred in the wild, rather than a signature for a multitude of distributed releases of a non-Wuhan-derived version of the virus (or a key element for the improved fitness of the B.1 model). This would require accepting that no convergently-evolved Wuhan-to-D614G variant was able to gain a lasting foothold due to clonal interference (when more prevalent versions of a genome have the upper hand in an asexual population despite a lack of increased fitness). It is plausible; but to me, a separate release for B.1 and B.1.1 after Wuhan clearly rates as as good, or better, an explanation.
All of the B.1.1 mutations, including C241U, appear in the late 2021 VOCs (except for Beta and Delta, which drops the B.1.1 Nucleocapsid mutation to return to the B.1 quartet) and, except for C241U, also appear in BA.0, the last LCA for the Omicron siblings.
“Ethical Skeptic” “China’s CCP Concealed SARS-CoV-2 Presence in China as Far Back as March 2018.” (2021, November 15, with updates.) theethicalskeptic.com
As far as I can tell, the analysis of “Omicron” as a descendent from a pre-Wuhan LCA still has not been updated to address BA.2’s co-emergence.
For example, Ethical Skeptic proposes to reduce the “budget” of mutations which need to be ascribed to “Omicron” (BA.1) by crediting 2 years of apparent mutations to Wuhan, leaving BA.1 with just under 4 years. In the case of BA.1, this would imply that BA.1 made ~40 mutations after 2018 (or a bit less). But then how is it that BA.2 acquired another 6?

Because Ethical Skeptic’s analysis depends on molecular clocking, these extra 6 mutations, representing a 15%+ increase in mutation rate between BA.2 and BA.1 (again, because he is only crediting 40 or less, not 60, to BA.1), pose a fatal problem.
Waiving this 6-mutation discrepancy away sabotages the entire argument for using genomic clocking as a “proof” of how old the Omicron siblings are to begin with.
Ethical Skeptic’s proposal, along with his insistence on the virus’s rigid adherence to the molecular clock, also inadvertently (because he posted it before BA.2 was in the news) pegged the division between BA.1 and BA.2 at roughly the same time of the sequencing of Wuhan, so that all post-BA.0 mutations occurred in the just-under-two years before both siblings were sequenced. This requires an explanation for why both siblings were suddenly subjected to such radically different selection pressures, why they were suddenly nearly inert for mutations to the Receptor Binding Domain, and (obviously) why BA.2 accelerated synonymous mutations vs. BA.0 whereas BA.1 only managed to add two.
It is far easier to just assume that lab conditions rendered irrelevant any real-world time constraints on the mutation rate.
See https://www.nicd.ac.za/omicron-lineages-ba-4-and-ba-5-faq/, hat tip to Modern Discontent since my own research / references are a disorganized jumble at the moment.
The Omicron lineage BA.4 was first detected from a specimen collected on 10 January 2022 in Limpopo. As of 29 April 2022, BA.4 has been detected in all provinces. […]
The Omicron lineage BA.5 was first detected from a specimen collected on 25 February 2022 in KwaZulu-Natal. As of 22 April 2022, BA.5 has been detected in Gauteng, Limpopo, Mpumalanga, KwaZulu-Natal, the North West, and the Western Cape.
Incredible posts like this is why I subscribe to your substack. Well done!
Are you familiar with the viral quasispecies theory? According to this theory, an infection is really an infection with many related viruses, which coexist in varying proportions. So if Ba.1 is predominant in the Qs, it mostly shows in sequencing and you may miss the lower prevalent ba.2 and ba.4 and ba.5.
But once immune landscape changes, other Qs members may gain higher prominence.
So, it is possible that they were all released as one quasispecies?
Substack is where the conversation went that used to take place in the scientific literature, I guess. It's reassuring to see such thinking and explaining going on, as it includes self-doubt and allows for future correction based on more data. When contrasted with the top-down edicts of recent years, it makes one realize how amazingly corrupt it's become, as predicted by President Eisenhower in his Farewell Address.