
Should variants actually happen? Pt. 3
SARS-CoV-2 is not a [highly mutagenic] virus.

The following continues from Part 1:

In Part 2, the generic stability of coronavirus genomes was explored. This sets the stage for discussing the mutation of SARS-CoV-2 “as a coronavirus” and in practice, as sequenced all across the world for the last 3 years.
And so (with apologies for a weekend away from my workstation that was more disruptive to this project than I anticipated), let’s get back to not-copyright-infringing-ly “boffing” those evolution myths!

(Also: “The VOCs were intrinsically better at transmission” has been added at the bottom of this post, in reply to discussion in the comments.)

Tony Honk’s Pro Skater 2: Coronavirus Edition
How should we think about SARS-CoV-2 mutation “as a coronavirus”? For this, we return to the “THPS” model of RNA virus stability.
This model was explained in Part 1. In short, as if RNA viruses were playing a video game in which the goal is to maximize the score for jumping around on obstacles with a skateboard, the “buttons” (RNA nucleotides) for the best “moves” (amino acids) to jump from game element to game element (host receptors, etc.) never change so long as RNA viruses are playing in a given environment; so the ones that replicate the best always have the same buttons.
An additional facet of the “game” here, is that the RNA molecules which encode the 30,000 or so “button-presses” for coronaviruses must actually take part in the physical process of the game. Now, immediately, I would like to make it clear that this will be true to an extent for all viruses and all forms of life. But it is still instructive to visualize just how much of the coronavirus life cycle hinges on functional / optimal physical interactions between its RNA molecule and corresponding tool-kit of proteins. It is a lot more complicated than a skateboard; it’s more like a 5-figure racing bike.
In the following lamentably ugly schematic, RNA-protein interactions during the replication cycle are colored by time (violet for early; red for late):
As it turns out, every mutation potentially alters physical interactions with the coronavirus RNA molecule and the host's and its own protein machinery. This is not merely a theoretical fancy; again, I would cite the example of the oral polio vaccine viruses. For two of the three strains, it was later found that the mutations responsible for attenuation were typically in the noncoding region of the molecule that corresponds to the “loop” structure in the cartoon above.1
Only one of these differences, a mutation from U to C at position 472 in the presumed noncoding region of the genome, is a back mutation to the wild type sequence. […] The nucleotide at this position is the only one which correlates directly with virulence, being uridine in the attenuated strain and cytidine in both virulent strains.2
The mutations deform the loop and lead to poor translation of the molecule in host cells (leading, immediately, to deattenuation3).
The OPV strain reversions serve as proof that a so-called “silent” mutation (one that doesn’t change anything about the proteins the RNA molecule codes for) can transform a tiger into a kitten, and vice-versa; and there isn’t even anything particularly astonishing about this fact.
Coronaviruses further evince extreme constraints on RNA composition in the form of “style features,” which include (partially) understood promoters for protein-interactions and modifications, high amounts of base-pairing, and a global prevalence of nucleotide repeats. In terms of the first item on that list, SARS-CoV-2 has a known “signature” (acgaac) that prompts reattachment of the RNA leader sequence to end-of-the-line genes, including spike and nucleocapsid, so that those genes can actually be read by the host cell. So almost any change to those segments will be fatal.
Regarding base paring, 62 to 67% of the RNA nucleotide “letters” in coronavirus genomes analyzed by Simmonds were “paired,” meaning they form a string that is predicted to ziplock together with a nearby string.4 Mutations to any of these paired nucleotides would potentially weaken those string-pairings by generating a conflict with the nucleotide in the opposite position (or at any rate, those mutations would alter whatever it is that leads coronaviruses to favor base-pairing).
Regarding nucleotide repeats, they are rampant, as previously shown in my text-format sequence upload for SARS-CoV-2:

Heavy use of nucleotide repeats correlates with some “odd” amino acid choices. For instance, since any codon with three uridines in it will code for a “F” (phenylalanine), SARS-CoV-2 proteins… have a lot of F’s (5.07% of all residues, vs. 3.65% in sequenced human proteins). This obtains in other coronaviruses as well.

To relate back to the “THPS” model of RNA stability, coronaviruses like to “jam the buttons” rather than move around between buttons rapidly. Careful and regular repetition of a’s and u’s might simply be the lowest-entropy way for an organism to achieve incredibly high rates of base-pairing; or contribute to gene regulating RNA-protein interactions described in my cartoon model, or all of the above.
Phenylalanine, by the way, is what your body metabolizes out of aspartame, the artificial sweetener. As an essential (not-made-by-your-body) amino acid that is a precursor to the molecules responsible for what your nervous system operatively uses as “energy,” some neural symptoms of coronavirus infection may simply result from the virus hoarding what is normally a “rare earth metal” in terms of your normal protein output, leaving your body depleted in derivatives like dopamine and adrenaline. I have no corroboration for this wild speculation; I am merely inserting it to depressingly remind readers how little we humans understand about anything.

It may further be the case that heavy use of “F” in coronavirus proteins places a drag on production of the same; or certainly that there will be many instances where the bulky “F” molecule could more optimally be replaced by a simpler, lighter alternative. And no doubt that when the virus shows up in a cell all dressed in weird aromatic rings, akin to a burglar showing up to your house in poodle-print pajamas, there are confused glances. But every reduction of an “F” would sacrifice a u-repeat, and potentially disrupt that giant molecular dance that the RNA molecule must perform every time the virus replicates. Really, that is all that I would like to point out here. Coronaviruses are really genetically constrained (as are all organisms).

Rounding back to my cartoon schematic above: Mutations that render proteins nonfunctional (including the proteins responsible for managing and modifying the RNA molecule in precise ways) might typically manifest a binary failure outcome, represented as a “N” as opposed to a “Y” for successful replication down the line (after the RNA molecule gets successfully transferred to a new cell by the functional proteins coded by the parent virus). Viral copies with these mutations are in an interesting limbo between useless debris and co-infecting “sacrificial pawn” status. Mutations that disrupt or interfere with RNA-protein interactions might impair replication on an analog scale, which I have simplified as “.1-.9” (assuming that differences below or above that threshold are functionally 0 or 1). Lastly, the “1” for this optimum will necessarily still involve a lot of molecular conflicts and compromises, with coronaviruses frequently modulating RNA secondary structure as a sort of perpetually unfinished symphony.5
This dynamic will be revisited in the final post as part of the argument that genetically hyper-novel “variants of concern” should not emerge as regularly as they have. Essentially all of the above will potentially be repeated; as I am uncertain just how to divide between background and foreground on this topic.
OK, let’s properly get back to our itinerary-defined myths:
iii. SARS-CoV-2 mutates a lot in transmission
From here, this post is being uploaded in a more-or-less “beta” version. I could do better to document the evidence for the virus’s low mutation rate; and may come back to edit and expand this version. But I think it is enough simply to throw a few quotes and pictures and random thoughts at the reader for now, “BMM Research” style. Ultimately, my goal in this series is to try to convey to the reader what I think several researchers have already observed by now, but have not been permitted to voice the logical conclusion which follows forth.
Early days
The genetic tranquility of SARS-CoV-2 was evident after the first wave, in 2020. From the same review previously quoted in this series, “A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants”:6
Most substitutions were found in a single sequence; only 8.41% (n = 2,474) of the polymorphic sites showed substitutions in two or more sequences (Fig. 1B). Only 11 mutations were found in >5% of sequences, and only 7 were found in >10% of sequences (3 of which were adjacent)
So, “variants,” as referred to in the title, were in those early days simply a description of the hoards of dead-end, “one-off” mutations that popped up in sequences. Reading backwards, the 7 mutations in question were those that defined B.1 and B.1.1; the latter of which is still the “starting point” for Alpha and the Omicron variants; the former of which is the starting point for Beta and Delta. Besides those two variations of the original recipe, no mutations “take.” So if Billy Exampleguy in New York City had his infection sequenced in March, 2020 and showed a c-ExamplePosition-u mutation, it didn’t show up any more down the line. Or at most it only showed up in an amount potentially consistent with random chance mutations happening in other people.
If most infections spread throughout the calendar stem from previous infections, in a chain, there should be a sort of nested, fractal pattern to “random” mutations. If B.1 accounts for a third of all infections (it was actually more than that; but as an example), then some mutation in B.1 should account for a sub-third (10%), and something in that another sub-third (3%), and so on; with obvious variability due to different transmission chains being provided different amounts of human “fuel” in the lockdown era.
So, I’ll be blunt here and state that the pattern of first-wave mutations described in the “candidate” paper is theoretically consistent with someone going all around the world and delivering B.1 or B.1.1 virus directly to human environments, with everyone getting their own discrete infection and not transmitting it. But, to this day (since first floating this idea in Omicron Origins) I remain ambivalent about the case here.
For one thing — the whole point about this entry in the series — coronavirus mutation isn’t “random.” The extremely poor “take” of most early mutations is instead exactly consistent with stabilizing (or “purifying”) selection7: Billy ExampleGuy's seemingly innocent random mutation was not able to survive because it messed something up. (This also extends to cases where mess-something-up mutations might still be molecularly frequent, resulting in nonbranched or convergent sequences.8) Therefore, going forward through time, the virus changes very little even if discrete infections can produce seemingly dynamic genomes. In a preview of the review of this topic that the next posts will deliver:

For another thing, the lockdown and gaps in sequencing in the first wave may have created some sort of bias for “first from root” cases; especially given that the cutoff for this paper was in April.
Further, in favor of natural spread in 2020, is that in between the “candidates” paper and the post-Alpha era, novel clades did begin to take root in various regions (those designated as “20C” to “20G” in Nextstrain); and these tended to register natural-looking “nested” sub-mutations of their own.9 This repeats again (at least) for Delta and the post-BA.1 Omicrons.
Additionally, the overall incidence of mutation observed in early 2020 (and afterward) is consistent with general mutation rates of coronaviruses. So, even if most of these mutations were dead ends, they still appeared (and then vanished) at the rate that would be expected for natural transmission.
But, this is still a huge problem
The current understanding of virus host species jumps, right or wrong, is that viruses do not “land” at optimum; instead of being punished, new mutations should initially be rewarded (in a step-wise manner, presuming that “random chance” hasn’t made it so that no improvements are possible in the new niche without crossing a fitness valley).
To quote from Simmonds, in a 2019 paper that strongly accords with the “THPS” model of RNA stability (emphasis added):10
In this Opinion article, we argue that the extraordinary conservation of virus genome sequences is best explained by a niche-filling model in which fitness optimization is rapidly achieved in their specific hosts. Whereas short-term substitution rates reflect the accumulation of tolerated sequence changes within adapted genomes, longer-term rates increasingly resemble those of their hosts as the evolving niche moulds and effectively imprisons the virus in co-adapted virus-host relationships. Contrastingly, viruses that jump hosts undergo strong and stringent adaptive selection as they maximize their fit to their new niche. This adaptive capability may paradoxically create evolutionary stasis in long-term host relationships. While viruses can evolve and adapt rapidly, their hosts may ultimately shape their longer-term evolution.
Another comment is provided by Neher, whose paper is discussed in the “And Beyond” section below:11
In addition to segregating deleterious mutations, early viral evolution after a host switch can also be driven by anomalously fast adaptation. A dramatic change in environment, e.g. a host switch, likely results in many opportunities for mutations that increase fitness. Such transient increases in the rate of adaptation are common in experimental evolution
As I am far from the first to point out, it is peculiar that SARS-CoV-2 didn't seem to experience these “growing pains.” However, there are caveats. First, I would note that most analyses leave out 3 likely mutations that occurred between the release of SARS-CoV-2 and Wuhan (“WH,” or Before Wuhan), the pre-mutation versions of which later appeared in sequences in China and Washington.12 Taken with B.1 and the even more radical B.1.1 mutation set, one might propose that the early period of the virus’s transmission in humans was more dynamic than it seems. In fact, this is exactly what Neher's analysis finds, with the most primative SARS-CoV-2 clades showing the highest rate of mutation.13
Secondly, if contrasted with whatever the rate of coronavirus evolution in the true long-term (as in, millions of years), the "slow" rate of mutations in SARS-CoV-2 might paradoxically be fast.
However, if not definitive, it is still absolutely arguable that what should have been observed after the appearance of this virus, were it truly “half at home” with its new host, is organic transition between rapid step-wise mutation toward a novel optimal, followed by stasis.

Instead, we have 7 potential step-wise mutations between 2019 and B.1.114 (which may simply be founder effect accidents; these were just neutral mutations that would be expected to become fixed by luck when the virus is just starting out), followed instantly by near-stasis, which is then suddenly interrupted by seismic shocks over and over.
A further argument is that even the Variants of Concern should individually show a honeymoon period of high mutations (discussed again in the And Beyond section below); instead, they are just (within their own clade) as flat as the trend.
In the Alpha Era
Nothing changed when “Alpha,” the first-to-be-sequenced “Variant of Concern,” landed on the scene in September, 2020 (it was not granted the greek letter name until the next May). As in the paper by Andrew Rambaut and collaborators (emphasis added):15
We found that the evolutionary rate of the ancestral branch was an average of 2.77 times higher than the background rate [Meaning: The accumulation of mutations between a B.1.1 “starting point” and Alpha, which somehow takes place (almost16) without any intermediary mutation-linkages showing up on any sequences anywhere, was way faster than the virus was mutating in general]
There is, however, little evidence for an increased rate of evolution within the B.1.1.7 clade itself: a regression [mathy line-drawing] of root-to-tip of genetic distances against genome sampling date (Fig. 1B) shows that the rates within the B.1.1.7 clade are very similar to those of the background lineage (4.6 × 10−4 and 4.3 × 10−4 nucleotide changes/site/year, respectively).

What makes Alpha aberrant is 1) That it has a lot of mutations, but still “takes,” as opposed to fizzling out, and 2) That it doesn’t have any of the endogenous mutations in the background — it doesn’t “come from” a version of the virus that was circulating in mid to late 2020 (instead, like the Omicrons, it is derived from a clean B.1.1 starting point), and 3) That no intermediary sequences between B.1.1 and Alpha exist or were ever found (this restates 2, and again is remarkable because 1 still happens).
And Beyond
The aberrance of Alpha, of course, was not a one-off event. Other sequences have “parachuted” into reality from a B.1 or B.1.1 starting point, including most notoriously the Omicrons. A preprint uploaded last month remarks on the paradoxical, or self-defying “trend”:17
The dichotomous pattern of SARS-CoV-2 evolution with step-wise [and slow] evolution within variants and atypical bursts of evolution leading to new variants has been investigated by Tay et al. (2022) and Hill et al. (2022) [the paper discussed above], who showed that the rate of evolution along branches giving rise to new variants is up to four-fold higher than the background rate.

Neher suggests that this difference is driven by some exponential, chain-reaction-like accumulation of beneficial mutations that occurs during the “cryptic” (lab, cough-cough — excuse me) development of the variants (in some magic immunocompromised star far, far away):
Nevertheless, all clades and variants are compatible with a single “back-bone” molecular clock that runs substantially faster than the “within variant clock”. This accelerated back-bone clock is likely driven by exponential amplification of beneficial mutations.
If this strikes the reader as plausible, allow me to argue otherwise. There is no self-evident force that would prevent a naturally-evolving variant from spilling “out of the cryptic evolution chamber” as it approached the optimal to which it was exponentially beneficially mutating toward. This very principle is why, so far as we know, mutation appears accelerated after host-switch: The host switch occurs when the virus becomes near a new optimal, not after it already arrives there. For this reason, we can see those “finishing touches” reflected in sequences (including well after the optimal version may arrive on the scene, as near-optimal versions would still have their active chains of transmission for some time).
As already mentioned, it’s at least possible that the transition of the original release to B.1 was just such a dynamic period, even if not that dynamic-looking in absolute terms.
So why don’t any of the “variants of concern” have a burst of initial mutations?
All of them, I would say — but really, at least some — should have experienced such an initial burst, if indeed there was anything about their mutations that improved fitness at all, as they would all have essentially operated as new viruses making a host switch. As an analogy; your iPhone will have small updates more frequently just after big updates (i.e. new OS versions).
And at least Alpha, which arrived in the heavily-sampled UK, and the Omicrons, and some kind of left-over trace of one of VOCs from less well-surveilled areas, should have left a record of this early mutation burst somewhere in the sequences.
Instead, the VOCs (as individual clades) seem to mutate more slowly as time goes on (vs. the rates within clades that preceded them).

There’s more to mine from the Neher paper; it will be revisited in the coming posts arguing that variants should not happen.
There are also more citations to offer commenting on the low rate of SARS-CoV-2 mutation, and contrast with VOC “builds,” apart from these. It is something that is being noticed. I may edit extra citations in later.
And, the next post, making the first argument that variants should not exist, will review the Trevor Bedford presentation that initially seeded the idea of “parachuting variants” in my head.
The final* myth:
iv. The “Variants of Concern” were immune escape-y

An obvious exception here is Omicron — especially “BA.0,” the version of Omicron that existed before BA.1 and BA.2 diverged into two different animals. Otherwise:
Variants of concern were not remarkable for mutations to antibody-targeted regions of the spike protein18; nor are such mutations what usually appear to “define” the infection advantage of the variant.
In the example of Alpha, Hill, et al. find that it outperforms a local strain that carries the exact same N501Y mutation, which is the only one Alpha has that is near the receptor binding domain:
N501Y in particular has been monitored throughout the pandemic, due to its ability to increase binding to the ACE2 of human and murine cells. However, it appears that by itself, it is not necessarily enough to create a VOC, as there was a cluster in Wales in late 2020 defined by N501Y, but without the NSP6 deletion, which was rapidly outcompeted by Alpha19
They further comment:
Finally, Beta and Omicron are the variants with the most evidence for immune evasion (Cele et al. 2021; Hu et al. 2022), and both variants share the common mutation K417N in the Spike protein (a similar mutation, K417T, is found in Gamma, Fig. 4C). K417N has been found to confer reduced susceptibility to neutralisation by specific monoclonal antibody therapies (Starr et al. 2021). This mutation also arose in AY.1, the so-called ‘Delta plus’ variant descended from B.1.617.2 (Kannan et al. 2021), but this variant did not appear to acquire any noticeable advantage compared to the background Delta wave (outbreak.info).
Those mutations that did appear, may correspond to high levels of immunity in the regions where the variants originate; e.g. India for Delta, Brazil for Gamma, South Africa for Beta and maybe BA.4/5 (the other immune-evading mutations reside in the BA.0 “ancestor,” and are thus relatively ancient). These could, in other words, be sub-variants of ancestor genomes that were lost before being sequenced (this would fit in the case of negatively selected, pre-immune-escape genes).
Regardless, those mutations did not confer immune escape to the variants in question in vivo. Natural immunity remained robust against Delta, Beta, etc., until the Omicrons.20 In the example of Brazil and Gamma, the variant appears to spark a rise in cases despite sky-high seroprevalence; but it doesn't actually cause many reinfections; in the early stages they were so rare as to be published one at a time:21
[I]n Manaus, Brazil, a study of blood donors indicated that 76% of the population had been infected with SARS-CoV-2 by October, 2020. […]
In this context, the abrupt increase in the number of COVID-19 hospital admissions in Manaus during January, 2021 (3431 in Jan 1–19, 2021, vs 552 in Dec 1–19, 2020) is unexpected and of concern […]
P.2 variants with the E484K mutation [i.e. Gamma variants] have been detected in two people who have been reinfected with SARS-CoV-2 in Brazil
But the hype over immune evasion in the case of Delta in particular even claimed myself; after all, I discussed (the myth of) “Original Antigenic Sin” in relation to Delta, even though Delta shouldn't actually result in very different antibodies.22
And so, 2020-class VOC spike mutations instead occur elsewhere, and have possible effects on other factors like transmissibility and pathogenicity (as do mutations outside of spike, naturally):
Beta increases binding to sialic acid, potentially resulting in transmission incompetence (due to SARS-CoV-2 lacking genes for the protein that helps re-release it from sialic acid).
Alpha reprises N501Y (the "mink farm" mutation) as does Gamma, and introduces the P681H mutation which potentially modulates nuclear import of spike, but no one knows yet.23
Gamma introduces H655Y, which is found to lower fusogenicity (it’s also in the Omicrons).
Delta introduces P681R, which dramatically heightens fusogenicity.
Etc. (“etc.” here means the other aberrant clades are less well-studied). And so one would be forgiven for wondering if the virus, in its mystical “cryptic” evolution chambers in myriad different, but seasonally synchronized chronically infected, immunocompromised stars, was, you know, “experimenting” with mechanically relevant spike protein mutations to then beam back to Earth for further observation.
You know, as viruses do.
*Not the final myth: An additional post discusses whether the “Variants of Concern” were intrinsically more fit than pre-VOC versions.
That Leaves… No Explanation
In sum (again, I could do a better job citing and laying out my case here), nothing appears to account for the simultaneous, seismic, but also abruptly self-limiting bursts of (“cryptic”) evolution that leads to the variants of concern. They appear in highly-immune countries and barely-immune countries at the same time; they don’t cause reinfections; they don’t follow the (semi-mythical) precedent of flu’s antigenic drift or shift.
They just… parachute in from nowhere.
Next: There Should Not Be Variants, Pt. 1.
If you derived value from this post, please drop a few coins in your fact-barista’s tip jar.


For type 2:
Valesano, AL. et al. (2020.) “The Early Evolution of Oral Poliovirus Vaccine Is Shaped by Strong Positive Selection and Tight Transmission Bottlenecks.” Cell Host Microbe. 2021 Jan 13;29(1):32-43.e4.
Three mutations—A481G, U2909C (VP1-I143T), and U398C—were inferred to be under the strongest selection pressure and precede subsequent substitutions. The A481G and U398C mutations are located in the 5′ noncoding region and are functionally important to RNA structures in the internal ribosome entry site (IRES). All three mutations are known molecular determinants of attenuation, occur within the first 2 months after vaccination, and are associated with increased virulence in animal models
Footnote 2 corresponds to type 3.
Cann, AJ. et al. (1984.) “Reversion to neurovirulence of the live-attenuated Sabin type 3 oral poliovirus vaccine.” Nucleic Acids Res. 1984 Oct 25; 12(20): 7787–7792.
Martinez, C. et al. (2004.) “Shedding of Sabin Poliovirus Type 3 Containing the Nucleotide 472 Uracil-to-Cytosine Point Mutation after Administration of Oral Poliovirus Vaccine.” J Infect Dis. 2004 Jul 15;190(2):409-16.
Simmonds, P. (2020 a.) “Pervasive RNA Secondary Structure in the Genomes of SARS-CoV-2 and Other Coronaviruses.” mBio. 2020 Nov-Dec; 11(6): e01661-20.
Hence why coronaviruses do not seem to share (have conserved) RNA secondary structures, suggesting that those structures mutate faster than other gene regulation factors. See (Simmonds, P. 2020 a.).
Dearlove, B. et al. “A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants.” Proc Natl Acad Sci USA. 2020 Sep 22; 117(38): 23652–23662.
Again, there is a semantic difference between the two, but in the “THPS” model it is not meaningful to distinguish between them.
Examples are the 5784, 10319, 21575, 28657 and 28887 C>U mutations that are not found to be monophyletic in the analysis by Simmonds:
Simmonds, P. (2020 b.) “Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories.” mSphere. 2020 May-Jun; 5(3): e00408-20.
This paper will be central to the next post.
Obviously, I am being completely non-mathematical here, in keeping with my “brute force” approach to phylogenomics. An example is 20E, which dominated Europe in the summer of 2020 and appears naturalistic in sub-mutations among 173,086 sequences.

Simmonds, P. Aiewsakun, P. Katzourakis, A. (2019.) “Prisoners of war - host adaptation and its constraints on virus evolution.” Nat Rev Microbiol. 2019 May;17(5):321-328.
Neher, RA. “Contributions of adaptation and purifying selection to SARS-CoV-2 evolution.” biorxiv.org
Kumar, S. et al. (2021.) “An Evolutionary Portrait of the Progenitor SARS-CoV-2 and Its Dominant Offshoots in COVID-19 Pandemic.” Mol Biol Evol. 2021 Jul 29;38(8):3046-3059.
As Nextstrain labels the Wuhan-Hu-1 sequence “19a,” I never stop feeling tripped-up about any of the literature on the “alpha” version which presumes Wuhan to follow. Note that the Zoonati have renounced this inferred order of mutations, because politically inconvenient.
However, this may also be a reflection of a defect in the RNA-copying protein (rdrp) which was potentially fixed by the P314L mutation in B.1, leading to higher within-clade fidelity.

I am including the 3 mutations to B, and counting N RG203KR as one mutation as it likely occurred in a single template switch event (or lab edit…).
Hill, V. et al. “The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK.” Virus Evolution. Volume 8, Issue 2, 2022, veac080.
As the paper discusses, there is one sequence that matches for four of Alpha’s mutations. However, this is poor corroboration for a natural origin as four matches among the entirety of pre-Alpha sequences might simply arise as a lucky “lottery pick” (akin to the highest winner of a lotto drawing having only four numbers right).
One way to visualize this is with Röltgen, et al.’s Wuhan-relative binding plots. Higher dots / bars denote that the variants in question are more immune-evading (with the red bar for natural infection being of the most interest, since the VOC’s emerge before the vaccines):
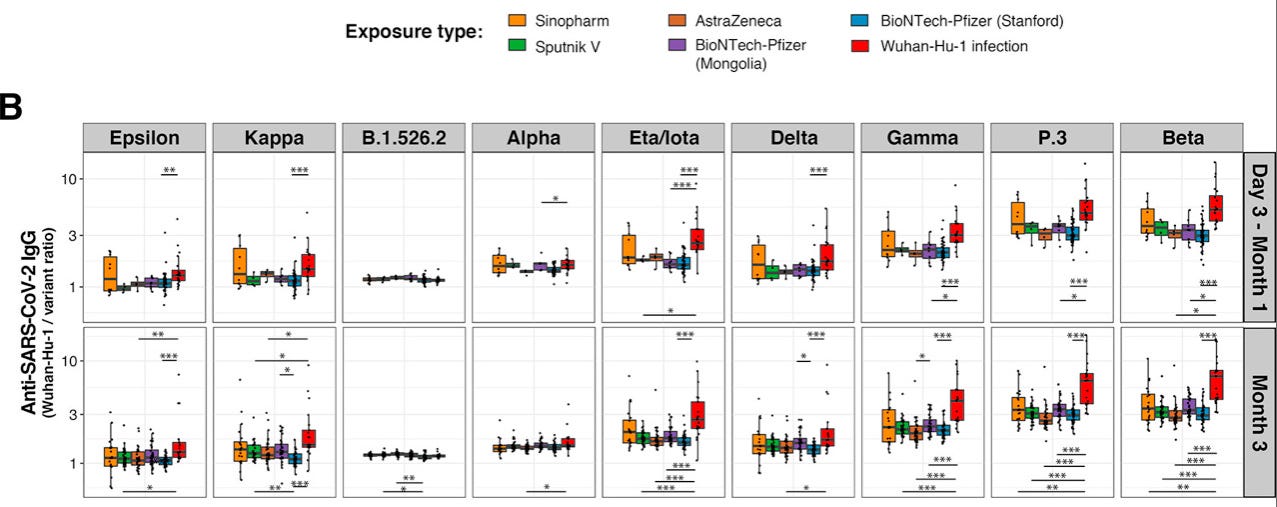
Hill, V. et al. “The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK.” Virus Evolution. Volume 8, Issue 2, 2022, veac080.
Goldberg, Y. et al. “Protection and Waning of Natural and Hybrid Immunity to SARS-CoV-2.” N Eng l J Med. 2022 Jun 9;386(23):2201-2212.
Sabrino, EC. “Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence.” Lancet. 2021 6-12 February; 397(10273): 452–455
Again, referring to “Even-Steven.”
See mark-up on my spike sequence map.
So, why doesn't the vax work? Apparently it does mutate and they're still distributing vax for variants that aren't in wide circulation. A competent medical industry could probably keep up, issuing new vax more frequently, but the insanely corrupt approval process precludes that, so we're stuck with the vax they managed to sneak through, regardless of ineffectiveness. That, and an ample supply of stupid people assures the gravy train continues a while longer, and whatever nefarious motives might be behind it. Maybe they'll find a vax for stupidity. Prosecuting the corruption will wise a lot of them up.
If the vast majority of intermediates (between variant lines) fall in fitness valleys, then as a null (vs. continuous lab update) hypothesis it would be helpful to be able to estimate the probability of crossing any of those valleys *within a single infected host* given the rate of replication errors and the total number of infections. For example, if - on average, and I'm just making this up - every infected cell produces 1000 virions with no genome changes, 100 virions with a single nucleotide changed, 10 with two changes, all the way out to 10 changes occuring in one out of every ten million infected cells, etc., then we could calculate the probability of new variants arising.
Also, would it be likely that the initial further single-nucleotide changes within a new variant that optimize fitness would occur within the first few hosts, thus causing the variants to *appear* to be stable by the time they are first sequenced?
Finally, it seems to me that - until Omicron - the new variants caused further waves of infection by evading innate, rather than adaptive, immunity - i.e. by infecting people that the previous strains were unable to successfully infect, rather than by reinfecting those who had recovered. It is not surprising to me that it is easier for a virus to find new hosts by increasing infectiousness (ability to overcome innate immunity), and that only once a majority of people have been infected does the more difficult approach of evading innate immunity become the best evolutionary move. That's not to suggest that Omicron or any of the VOCs *weren't * lab releases - only that the observed pattern seems consistent with evolutionary pressures to me, and that (in the absence of abundant sequence data to track and analyze genetic changes) the overall progression of waves of infection and eventual reinfection doesn't seem especially anomalous for a new virus entering the human population.