
(Refining the) "Deep State Origin" case
Further defending the evidence for a synthetic origin, and expanding on implications of same.

Reader warning: This is nothing more than a draft of a new tentative theory for how the SARS-CoV-2 origin puzzle pieces might all fit together. But because full-length genomes and large data sets are still beyond my ability to crunch, I can’t verify whether there are any arm-socket conflicts in the suggested arrangement. Call the whole thing an elaborate fiction, if you would like.

Intro: Maybe the Whole Thing Was a Giant Troll
SARS-CoV-2’s “synthetic fingerprint” is both typical and aberrant. This makes the case for a synthetic origin seem weaker than I initially took it to be in my review of the bombshell preprint two weeks ago.

But this isn’t quite the whole story. Because in fact, the aberrance of this virus’s synthetic fingerprint makes both synthetic and zoonotic origin less plausible at the same time (more below); which means the balance does not necessarily change.
Instead, what results is that the required theory of mind for a synthetic origin becomes more complex; seemingly quickly veering into “Donald Trump’s Secret 3D Chess Plan to Purge the Pedophiles Any Day Now” territory. Whereas no matter how unlikely a zoonotic origin becomes, Nature’s budget for whimsy is infinite.
And so finally, stewing in thought at this unsatisfying impasse all day yesterday, I began to imagine the possibility that this virus’s creation was a true rogue agent act; a massive troll on humanity committed with shoestring resources by some nihilist nineteen year-old in a lab in southeastern Asia somewhere.
Perhaps, indeed, SARS-CoV-2 is that thing; but mostly, this way of looking at things lent to a rethinking of the case for a “US deep state” origin of the virus, and finding it not to be as complexity-burdened as it seemed.
Why BsaI and BsmBI Are Aberrant; Except Not
(Note, November 10: The discussion of the argument that SARS-CoV-2’s restriction site map is “random” or non-unlikely has been reformulated to take a more equivocal approach. The original text is in the footnotes.1)
The mistake I made when reporting and discussing the findings of Bruttel, Washburne, and VanDongen was in apprehending their choice of “chop here codes” (restriction sites) to look for in SARS-CoV-2 to be based on “chop here codes” added to previously published DNA<>coronavirus / “infectious clone” constructs. I can partially blame the fact that I was out of office; it’s difficult to compile stuff without a large display.
Here, again, the paper in question:

Now that I have compiled previous coronavirus DNA<>virus constructs, I realize that most of them, especially those published in association with Baric or the Wuhan Institute of Virology, use BglI (as, in fact, acknowledged in Bruttel and co.’s rationale).
For example, in 2017 when Shi, et al. finished rounding up another crop of bat-poop-recovered coronavirus genes and wanted to plug-and-play their individual spike proteins into the previously stabilized rWIV construct, they chose BsaI and BsmBI to splice in alternate spike sequences, but all the other junctions of rWIV were still BglI.

More ambiguously, when Baric’s lab quickly and quietly created their own DNA<>virus construct based on the SARS-CoV-2 genome in 2020, in order to try to mouse-adapt it “because reasons,” no mention of the restriction sites employed was ever made.

And so overall, while the argument in the “synthetic origin” paper is still strong, it doesn’t seem clear that the choice of BsaI and BsmBI for their SARS-CoV-2 restriction site map has an a priori “to avoid losing power” justification after all. Bruttel, Washburne, and VanDongen, simply remark, “However, while MERS and SARS have several suitably located BglI sites in their genomes, the close relatives of SARS-CoV-2 have only one conserved BglI site that is inconveniently close to the beginning of the genome.”
If we take this rationale as insufficient, it leads to two of the most oft-quoted weaknesses of the paper. The first is that the reader no longer has any reason to accept these results as not a manifestation of “p hacking.” The second is that the actual, end BsaI / BsmBI placement that makes up SARS-CoV-2’s restriction site map is predominately determined by apparently wild (natural) coronavirus genes, especially those published earlier this year in the “BANAL” sequence dump, from Laos:
Both these two problems, however, turn out to partially neutralize each other: The existence of a wild sarbecovirus backbone with a near-ideal BsaI/BsmBI map creates the justification for using these two restriction sites to score SARS-CoV-2’s map as synthetic or not: It makes sense that any worker converting BANAL to a DNA<>virus construct would leverage the near-ideal BsaI/BsmBI map already available in the genome. Here I am only restating Bruttel, et al.’s own remark: “However, while MERS and SARS have several suitably located BglI sites in their genomes, the close relatives of SARS-CoV-2 [i.e., the BANALs] [are pre-disposed for conversion to a BsaI/BsmBI-based platform].” It is still “p-hacking,” but only in the sense that anyone working with a BANAL-like backbone would surely look for whatever endonuclease combination the genome was already close to ideally mapped for.
How Likely is That Site Map in the Window?
Here is where the critics of the synthetic origin paper, on twitter, have made the claim that SARS-CoV-2’s conversion from near-ideal to ideal is unremarkable; it could obviously have happened by random chance. As my annotations show, only a few changes are required from the published BANAL genomes to arrive at SARS-CoV-2’s “construct-ready BsaI/BsmBI-map” (or for an ancestor to the sequenced BANAL’s to have been there all along).
“There are a couple obvious reasons why this stretches the zoonotic origin theory thinner than it was to begin with” — or so I said in my original version of this section. In fact the plausibility of a wild BANAL or BANAL-related virus having converted to an ideal BsaI/BsmBI map by chance is a more complex, fun problem than I realized. Before discussing the problem, I want to be sure to re-frame what the synthetic fingerprint paper is and is not. There are three categories of meaning (philosophy term) intrinsically suggested by this debate; however, I do not actually want the reader to understand all three as being defended here in absolute:
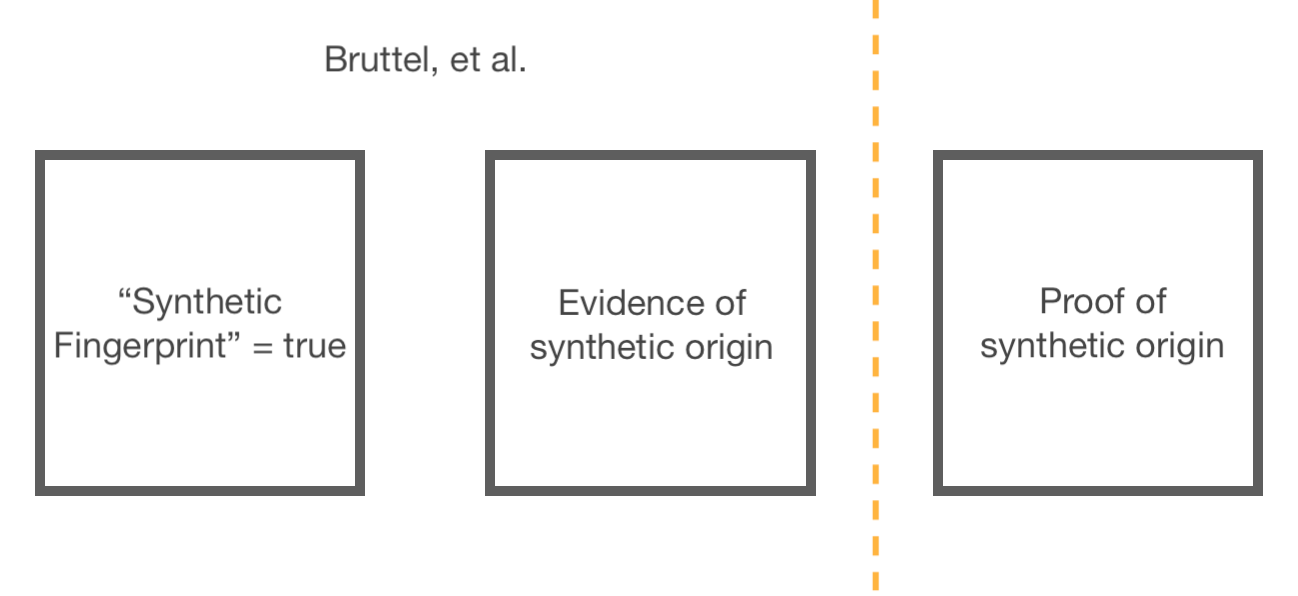
Regarding box 1, it is just a fact that SARS-CoV-2’s BsaI/BsmBI map is a “synthetic fingerprint,” however uncomfortable that fact may make the Zoonati. When humans create DNA<>virus constructs, it leaves marks that look like SARS-CoV-2’s BsaI/BsmBI map. In the same way that, if Elvis’s face appears on a potato, it may not indicate cosmic Elvis visitation, but the there-ness of the face is a simple fact which is indifferent to the validity of any other implications.
The validity of box 1, however, intrinsically grants validity to box 2. The synthetic fingerprint is evidence of a synthetic origin. But, as would be true in any court case in which forensic evidence was being cited to allege a human act, the question of whether the evidence is (by itself) proof of a synthetic origin rests on nothing so much as the likelihood of said evidence “just occurring” by coincidental chance. I am only asserting the second box to be a given. My argument regarding the third box is subject to intrinsic limitations on human understanding. I am not advocating certainty; given that the synthetic fingerprint evidence does not in fact exist “by itself,” but is one corroborating element among many, certainty on this point isn’t really important.
And so regarding the claim that this fingerprint could have arisen from the near-ideal BANAL maps by chance, it is first the case that being near-ideal does not, in fact, make it likely to randomly achieve an ideal. However close a random sequence gets to the ideal in question, the more likely it is that random mutations will take it further, not closer from the ideal. An intuitive example is any lottery-type sequence. Say your randomly-generated draw of 9 numbers from 1 to 4 is only one digit away from the winning draw:

Well, shouldn’t you be able to get the winning sequence just by shooting some more random changes at your own draw? No: Random evolution, it turns out, won’t be much help in getting you to the winning sequence. Statistical diffusion (my own phrase) makes it likely that only 1/36 of the next possible random changes will win; 3/36 will preserve your current near-ideal-ness; and the other 8/9 will take you further from the ideal. Within a few moves you will be unrecognizable compared to the ideal.
The limit, here, is that a BANAL progenitor is adjacent not just to one, but multiple possible “winning” site maps that would match a synthetic fingerprint.

As the above markup probably totally fails to show, there are numerous alternate move-paths to a “winning” site map (one with only five retained sites, but no increase in length of the longest segment); and numerous moves that would forgo “winning” (add sites, or reduce sites at the cost of increasing the length of the longest segment). In particular were a BANAL progenitor to “randomly” lose either of 17,320 or 17,963 (which surround the putative “D” stub), multiple other sites could be retained without failing to “randomly” achieve a synthetic fingerprint.
How, then, should the jury score the likelihood of a wild BANAL relative randomly having a fingerprint-matching map of some kind — and the resulting supposition that SARS-CoV-2 is that random map-haver? The question is more complex than can be accurately quantified. It would, for example, have to take into account different “budgets” for safe mutations in different parts of the coronavirus genome, as well as the undetermined safety of discrete site map mutations.
On the other hand, if such knowledge were in fact attainable, it may lead to an even better argument that wild achievement of SARS-CoV-2’s map is not even possible (it may be that some of SARS-CoV-2’s site losses would coincidentally confer low fitness for a bat coronavirus, or that they do in fact confer low fitness in SARS-CoV-2; a fact which would not matter much in the context of sustained, multiple releases).
It may also be the case — depending on how many safe site-additions are possible in the BANAL genome, which may not actually be any — that the failure of SARS-COV-2 to add any “novel” sites relative to the BANAL sequences is, paradoxically, better evidence of unlikely random mutation than novel sites would be.
Suffice it to say, that the assertion that SARS-CoV-2 could have “randomly” achieved a synthetic fingerprint map from a BANAL baseline, in bat-to-bat transmission in some un-sequenced wild virus — that such a pattern is likely or unremarkable — is baseless. My guess is that the entropy of random chance pushes BANAL away from the ideal to which prior random chance has brought it close to.2 It would require either natural selection — to pick closer-to-winning tickets out from the genetic swarm one by one, without committing any fatal changes — or human intention.
The other problem with using BANAL to dismiss a synthetic origin for SARS-CoV-2 is that it only magnifies the pre-existing problem with blaming the wet market: The BANAL genomes are closer to SARS-CoV-2 than those harvested in southwestern China; but they are found in northwestern Laos. The “zoonotic miracle” of how a yet-un-sequenced wild bat virus ended up spilling over to humans in Wuhan without doing so anywhere geographically between its natural reservoir and the wet market only magnifies by 200 hundred more miles and an international border. They do have humans in northwestern Laos, after all.
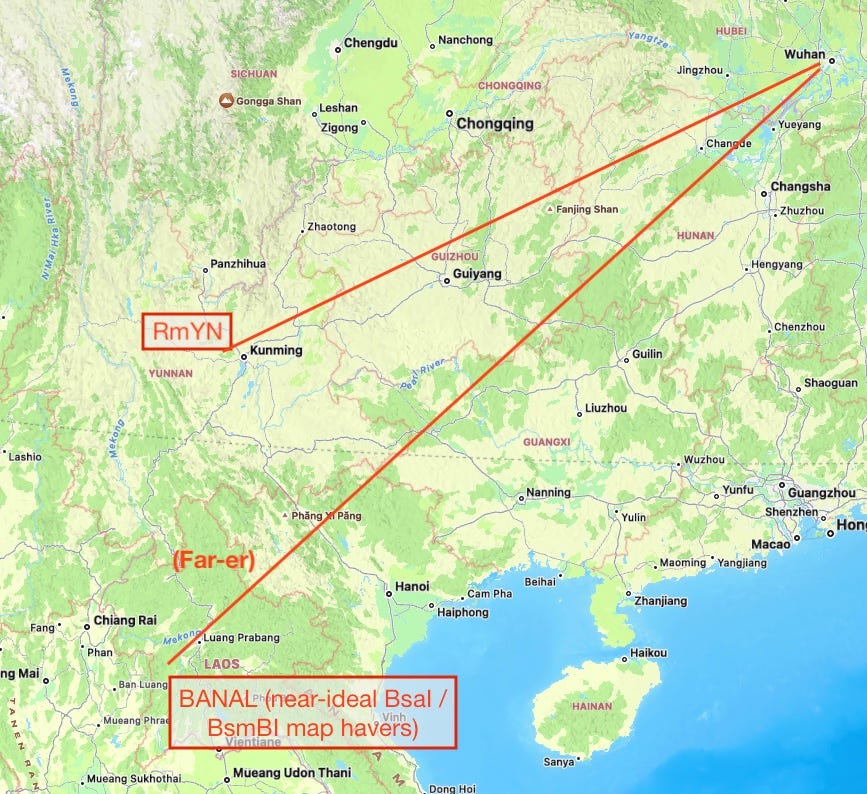
In embracing BANAL as an argument against the synthetic origin of SARS-CoV-2, in other words, the twitter Zoonati have only advanced the case for the implausibility of a wet-market-epicenter zoonotic origin. Bruttel, et al.’s reasoning that SARS-CoV-2 should be scored according to their BsaI/BsmBI map, because that is what a human worker wishing to convert BANAL to a DNA<>virus construct would build off of, is thus still compelling on balance.
Implications: So Does This Mean All Prior Published Work on Infectious Clones Is Out as a Clue to Who Made SARS-CoV-2?
But where I struggled, at first, in dismissing the “BANAL is close enough to ideal” argument is that embracing the evidence that a BANAL background was engineered into SARS-CoV-2 requires throwing out all known prior work associated with the Wuhan Institute of Virology and the Shi - Daszak - Baric axis. They are not known to have been working on genomes lending to a BsaI/BsmBI-mapped DNA<>virus construct.
So what was one supposed to imagine had happened, instead? As I mention in my introduction, my initial feeling about embracing the BsaI/BsmBI map was that it made the required network of actors and theories of mind too complex. I suppose I was attached to the notion that I could personally be inclined to believe in an “intentional US bioweapon”-type theory, but still refer to published work on Infectious Clones as supporting evidence. SARS-CoV-2 as a Propriety BANAL Construct felt like a leap into pure imagination: It required assuming, for one thing, creativity on the part of the actors behind SARS-CoV-2. All prior work on SARS-like viruses collected in China was abandoned, and a Cinderella virus from Laos was used instead.
At first, the only scenario that seemed to justify such a leap, in my mind, was that some rogue actor with a small lab had gotten lucky. But it was when I tried to imagine whether such a “trollish, for the lulz” origin for SARS-CoV-2 could lead to the same global farce and political, liberal disaster that has obtained for the last three years — when I perceived that this sequence of events was plausible — I suddenly realized that a Propriety BANAL Construct fits in with a US bioweapon theory without requiring any extra parts at all.
If Wuhan was already an ideal center stage for a misinformation theatre combining DEFUSE, Gain of Function, and Self-Fulfilling-Zoonosis-Prophecy, there would be neither a need nor an advantage to sourcing the genome for SARS-CoV-2 from Chinese samples. Why not quietly start from scratch in the Indochinese Peninsula, with some yet-unpublished bat viruses? The intrinsic unwieldiness of genomes — including 30 kilo-base coronavirus genomes — would ensure that any biolab working on coronaviruses at the site of the first acknowledged cases would not be able to forensically self-assess its own innocence quickly or confidently enough to avoid unintentionally covering up the same.
On the other side of the Pacific, while a Propriety BANAL Construct raises myriad questions about who is in the know and who is not — it remains obvious that Daszak has always been in the latter category, but becomes unclear which one Baric belongs to — raised questions are not the same as required assumptions. The model where the WIV theatre has nothing to do with SARS-CoV-2’s actual creation, I came to realize, does not require any more parts than a model in which actors are somehow working through the WIV, only to leverage the WIV as part of the theatre. In fact, it may be more conservative.
Meanwhile, as with any model, the structure of an “op” to frame an official zoonotic narrative and official “lab leak” counter-narrative via media and political plants or puppets, resulting in two “narrative factories” to distract from intentional release, remains self-evident:
Further rebuttals of the common Zoonati counter-arguments to the synthetic origin paper are included in the appendix.
Related:
(The DEFUSE document dump does not not look like an op.)
(Clarity on the timing of SARS-CoV-2’s movements in 2019 via blood samples in England.)
(The case for an Omicron Lab Origin.)
Appendix: Addressing the Arguments Against the Fingerprint Argument
As compiled by twitter user @flodebarre:
1 Evolution
SARS-CoV-2's BsaI/BsmBI restriction sites are consistent with expectations from evolutionary theory. They do not look anomalous.
1a) Kristian G Anderson: It is clear that SARS-CoV-2’s restriction map, when compared with a more relevant set of coronavirus genomes, reflects random noise.
Addressed above. Existence of near-IC-ideal maps does not make SARS-CoV-2’s possession of an ideal map either “random” or likely-to-occur-randomly. Even if a recombination is sufficient to achieve the ideal “small-largest-segment” spacing, it does not declutter the extra restriction sites. Small-largest-segments are a common “noise” artifact in high-RS-count maps, not low-RS maps. (This argument also more or less presumes that humans don’t really make Infectious Clones, and thus recognizing the fingerprint of Infectious Clones is inherently misleading; but humans do make Infectious Clones.)
1B) Alex Crits-Cristoph: “The “unusual” sites are all *exactly* found in natural bat coronaviruses.”
This is a fair criticism in so far as Bruttel, et al. perhaps place too much emphasis on the non-synonymous nature of the nucleotide changes that distinguish the SARS-CoV-2 BsaI/BsmBI sites from other sequences. Again, a counter-argument that takes human construction of Infectious Clones as a serious possibility is that known silent mutations are even more ideal than experimental silent mutations, as they have a higher likelihood of temperate secondary RNA structure effects. For example, rather than a wild recombination to explain SARS-CoV-2’s possession of 103/236/52’s 5’-region site motif, it is plausible that this motif was “borrowed” to complete the RS map in a 116/247 backbone (including one with a RpYN06-recombined Orf1). Either way, this argument is a complete strawman since the synonymous loss of mid- and 3’-genome BsaI/BsmBI sites is “unusual,” in the context of the wild BANAL genomes.
1c) A final tweet claims that RpYN06 has the same “silent mutations.”
This tweet is in fact only listing potential genes in RpYN06 that could become BsaI/BsmBI sites, which is completely besides the point of distinguishing human editing from random mutational noise, both of which build off of “almost-states” and prefer synonymous mutations to potentially fatal non-synonymous mutations.
2 Statistics
2a) It’s P-hacking!
Answered above. A P-hacking claim might have been rational before the publication of the BANAL sequences; since these are the plausible origin or origin-relatives of SARS-CoV-2 no matter what, it makes sense to analyze SARS-CoV-2 according to the near-ideal BANAL BsaI/BsmBI maps. The unlikelihood of a natural attainment / appearance in SARS-CoV-2 of a true ideal BsaI/BsmBI map, given no wild selection pressure for the ideal, remains.
2b) The authors only did their stats on the length of the longest fragment!
This argument doesn’t seem to understand why length of longest fragment is the most important quality control consideration from a DNA<>virus construct perspective.
3 Molecular biology
3a) Friedemann Weber: Seamless cloning can be used to avoid incorporating restriction sites into the virus’s genes.
Irrelevant, as can be done differently does not mean would be done differently. Published coronavirus DNA<>virus constructs embed the recognition sequences into the virus’s genes, not into an adjacent adapter sequence, as in rWIV. Once again the now-published BANAL sequences make leveraging of in-sequence BsaI/BsmBI sites the ideal for a human-use BANAL-based construct, regardless of whether one believes the synthetic origin argument or not. The paper Friedemann quotes clearly had trouble with their doing-it-the-hard-way approach, having to combine synthetic and PCR-amplified segments and verify rescue with, what-else, edited signatures, so his assertion that seamless is “standard” or “easier” is just rhetoric.
A further note regarding the use of visible restriction sites:
Even if seamless cloning is not in fact “standard,” one might propose it as more rational in the context of an “intentional release” theory-of-mind. However, such a proposal would assume that small-scale lab tests could predict how a DNA<>virus construct would in fact behave in the wild. Since such real-world information is only attainable after release, it is hazardous from a quality standpoint (besides harder to scale) to add a another “clean-up” step to the genome before release. For example, if the virus seems infectious in the lab, and then fails to spread in the wild, how would one trouble-shoot between the version studied in the lab and the changes made to “hide” the restriction sites? Any real-life quality control approach would favor leaving the restriction sites in the released product so real-world performance corresponds 100% to the lab-stock genome (the advantage of a DNA<>virus construct to begin with is not just to be able to apply target mutations, but to have a fixed, master copy of viral genes that isn’t subject to the “subjective” nature of passage distortion).
3c) SARS-CoV-2 fragment lengths are not 100% rational (the “D” stub segment).
This ignores that other published constructs (again including rWIV) have small segments as well. Not all endogenous restriction sites will be amenable to removal in practice, due to secondary-structure considerations, etc.


4 People
This section is literally dedicated to ad hominem attacks on Bruttel, et al., “to understand where these authors are talking from”…
If you derived value from this post, please drop a few coins in your fact-barista’s tip jar.


Original text:
The second is that the actual, end BsaI / BsmBI placement that makes up SARS-CoV-2’s restriction site map is predominately determined by apparently wild (natural) coronavirus genes, especially those published earlier this year in the “BANAL” sequence dump, from Laos:
[…]
Here is where the critics of the synthetic origin paper, on twitter, have made the claim that SARS-CoV-2’s conversion from near-ideal to ideal is unremarkable; it could obviously have happened by random chance. As my annotations show, only a few changes are required from the published BANAL genomes to arrive at SARS-CoV-2’s “construct-ready BsaI/BsmBI-map” (or for an ancestor to the sequenced BANAL’s to have been there all along).
There are a couple obvious reasons why this stretches the zoonotic origin theory thinner than it was to begin with.
First of all, being near-ideal does not, in fact, make it likely to randomly achieve an ideal. (*edit, November 8: The following section needs to be rewritten to account for the existence of multiple possible ideals; rather than assume SARS-CoV-2’s is the only one. So the math is a bit more fluid.) However close a random sequence gets to the ideal in question, the more likely it is that random mutations will take it further, not closer from the ideal. An intuitive example is any lottery-type sequence. Say your randomly-generated draw of 9 numbers from 1 to 4 is only one digit away from the winning draw:
Well, shouldn’t you be able to get the winning sequence just by shooting some more random changes at your own draw? No: Random evolution, it turns out, won’t be much help in getting you to the winning sequence. Statistical diffusion (my own phrase) makes it likely that only 1/36 of the next possible random changes will win; 3/36 will preserve your current near-ideal-ness; and the other 8/9 will take you further from the ideal. Within a few moves you will be unrecognizable compared to the ideal.
The math for the ~9 sites that make up either the ideal or off-ideal SARS-CoV-2 / BANAL maps is the roughly same, though the generic extra safety of synonymous mutations over nonsynonmous may reduce the realistic number of available next moves to something between 18 and 36. On the other hand, none of the near-ideal sequences are only one mutation away from the SARS-CoV-2 map; all of them require more than one successful toward-ideal mutation.
But since “winning” a clone-ready restriction site map is not favored by natural/wild selection pressure, it is not actually likely for the BANAL “cloud” genome to produce SARS-CoV-2’s map by chance; being near-ideal makes random mutation toward ideal less likely. And winning one toward-ideal mutation makes further toward-ideal mutations more unlikely. The entropy of random chance pushes BANAL away from the ideal to which prior random chance has brought it close to. It would require either natural selection — to pick closer-to-winning tickets out from the genetic swarm one by one — or human intention.
The other problem with using BANAL to dismiss a synthetic origin for SARS-CoV-2 is that it only magnifies the pre-existing problem with blaming the wet market: The BANAL genomes are closer to SARS-CoV-2 than those harvested in southwestern China; but they are found in northwestern Laos. The “zoonotic miracle” of how a yet-un-sequenced wild bat virus ended up spilling over to humans in Wuhan without doing so anywhere geographically between its natural reservoir and the wet market only magnifies by 200 hundred more miles and an international border. They do have humans in northwestern Laos, after all.
The best argument for this is already in Bruttel, et al. to begin with. From any “starting point” at an outlier low-longest-segment relative to number of segments position, further random mutation will push the genome in question toward the median.

To again speak in the language of games, what is being shown on this map is akin to a sample of chance-based game outcomes. Each of the analyzed restriction site combinations, when “dealt” through the genomes of coronaviruses, picks some winners in terms of low-longest-segment length relative to low number of segments. But as those winners continue playing extra hands (by mutating randomly), their fortunes will sink as surely as a human playing Windows Solitaire for more than five minutes at a time.
Axiomatically, the spread of outcomes above cannot occur unless being close to ideal makes further approaching the ideal less, not more likely. Therefor, the wild BANAL map is not likely to have achieved the ideal via random mutation.
I don't see why they can't have a library of potential bioweapon candidates from samples taken all over the world. There seem to be a lot of DTRA labs globally. I seem to remember clusters in SE Asia.
It is very clever to make bioweapons while funding similar research in a competitor state to use as a patsy. You would never farm out your actual best candidate virus research to a competitors lab however. I think Daszak's Wuhan lab work was the parallel construction and that is what the CIA recruited him for. I believe Huff says that the recruitment involved the CIA being very interested in the work that was being done with the Wuhan lab. Of note is that Shi Zhengli was an author of a research paper showing HIV-like characteristics of SARS-COV-2 published earlier this year. Why spill the beans about the special characteristics of your own nasty little bioweapon?
Talking up the threat of zoonotic spillover while making weapons from animal samples is also clever narrative preparation. The icing on the cake is that they blame human encroachment on nature and get to push a green message at the same time. Deindustrialize and depopulate or we'll keep releasing bioweapons that we blame on nature!
I’m sending this around today: https://reason.com/volokh/2022/11/07/november-7-as-victims-of-communism-day-2022/